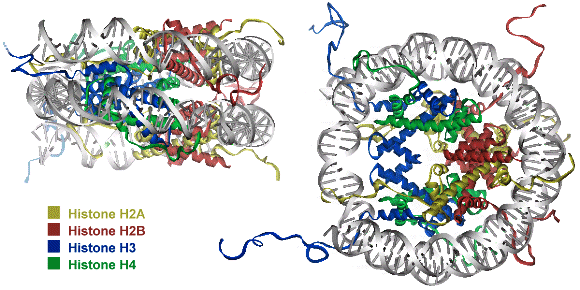
Chapter 1: Introduction Thesis Henry Jonker: PC4 and VP16 Niets is zo rijk als de onuitputtelijke weelde van de natuur - R.W. Emerson General introduction to RNA Polymerase II transcription and regulation by the VP16 activator and PC4 cofactor Introduction Understanding the connection between genotype and phenotype is an ongoing effort in molecular biology. This requires fundamental knowledge on gene expression and in particular on the transcription process. Biomolecular studies have provided an enormous amount of functional and structural information on the interaction of the central molecules of life: DNA, RNA and proteins. It has become clear that the gene transcription machinery is a complex process that requires the interplay of many transcription factors (Fig. 1). DNA is organized into chromosomes, each packed into chromatin. The minimal unit has been defined as the nucleosome. Activators, bound to specific DNA enhancer sequences, can recruit chromatin remodeling complexes and modifiers that destabilize the nucleosome to access the promoter DNA sequences. With support of cofactors, the activators can recruit the basal transcription factors and RNA Polymerase II to the nucleosome free regions in order to assemble a pre-initiation complex, needed for transcription. Various interactions between these factors have been identified, but still little is known about the detailed molecular mechanism by which individual genes are activated. This chapter will focus on the current findings on the chromatin complex and the RNA Polymerase II transcription machinery. The packing of DNA into chromatin and the regulation of its structure by chromatin remodeling factors and modifiers is presented first. Subsequently, an overview is presented of the structural and functional aspects of the basal transcription factors, needed for RNA Polymerase II transcription. At last, the activators and cofactors are discussed, with a special focus on the transcriptional activator VP16 and the general transcription cofactor PC4, which are the subject of this thesis. 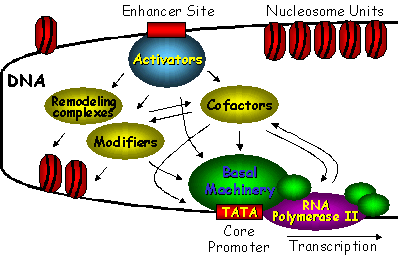 Figure 1. Schematic overview of the interplay between several classes of proteins that cooperate to initiate transcription of DNA. The chromatin remodeling complexes and histone tail modifiers destabilize the nucleosome in order to make the DNA accessible. Activators recruit the chromatin remodeling complexes, modifiers, cofactors and basal transcription factors in order to access the DNA and assemble the pre-initiation complex on the core promotor. Many cofactors are required to mediate signals between transcriptional activators and the basal transcription machinery. Nucleosomal DNA (de)compaction in the Chromatin complex Even in the most simple organisms, DNA molecules are very long. The total human genome appears to be over 3 billion base pairs long and contains about 30.000 to 40.000 protein-coding genes (1). In order to fit about 2 meters of DNA into the nucleus of an eukaryotic cell, the chromatin structure is composed of repeating compact nucleosome units that consist of about 157-240 base pairs and a histone octamer complex (2, 3). The histones have been designated H1, H2A, H2B, H3, H4 and H5. Around 145-147 base pairs are tightly wrapped around the histone octamer that consists of a central histone (H3)2(H4)2 tetramer and two histone (H2A-H2B) dimers at the start and end of the DNA path (Fig. 2). In addition, the so called linker histones (H1 and H5) are bound on the outside of the nucleosome. These linker histones are involved in chromatin coiling and folding and protect an additional 20-22 base pairs of DNA (4). Each of the histones has a characteristic histone fold and an unstructured tail. The compact histone fold is based on a long central helix with two shorter a-helices and DNA connecting loops. The histone-tail is subject to modification, which influences gene activation and interactions for higher-order (hetero)chromatin folding. Since DNA is compacted in the nucleosome, its accessibility for transcription is mostly limited to linker DNA that connects the core particles. The repressive effects due to chromatin packaging are mostly due to histone-DNA interactions in the nucleosome and histone tail interactions in the chromatin fiber. Entire genes can be inactivated (silenced) by the formation of a condensed heterochromatin structure. Chromatin repression can be alleviated by ATP-dependent chromatin remodeling complexes and by modification of the unstructured histone tails (5-8).
Histone-tail modifiers The histone tails are subject to many types of modification, such as acetylation, phosphorylation, methylation and ubiquitination. Histone acetyl transferases (HATs) and histone deacetylases (HDACs) regulate the histone-DNA and inter-nucleosomal histone-tail interactions by neutralizing the positively charged lysine residues. Acetylation of the histone tails destabilizes the nucleosomal structure and increases access of transcription factors to the nucleosomal DNA. In contrast, deacetylation of histones is important for formation of the heterochromatin structure and causes transcriptional repression. Modification of histone tails by means of phosphorylation and dephosphorylation is an important regulatory mechanism for cell division and transcriptional activation (9). H3 phosphorylation correlates with proper chromosome condensation and segregation during mitosis and meiosis (10, 11). Histone modifications may influence one another, as the phosphorylation of S10 in H3 stimulates the efficiency of acetylation (12, 13) and consequently activates transcription. Interaction of different histone modifications is further demonstrated by the repression of H3 S10 phosphorylation by H3 K9 methylation (14). Methylation of the core histones is catalyzed by histone methyltransferases (HMTs) and can occur on lysine and arginine residues and is associated with transcription regulation and heterochromatin formation or gene silencing (15, 16). Distinct methylation of H3 K9 provides a critical mark on the histone tail, leading to recruitment of the heterochromatin protein 1 (HP1), which mediates gene silencing (17, 18). In contrast to acetylation and phosphorylation, methylation does not alter the overall charge of the histone tails, but it increases the basicity and hydrophobicity. Lysine K9 of the H3 tail can be targeted for either acetylation or methylation (19). While heterochromatin assembly is negatively regulated by acetylation of the specific lysine in H3, it is stimulated by methylation of this residue, suggestive for a regulatory mechanism that may be controlled by phosphorylation of the neighboring serine. Specific histone methyltransferases have been shown to directly interact with RNA Polymerase II and are suggested to be involved in regulation of initiation and promoter clearance (20). Finally, transcription and chromosomal segregation are affected by ubiquitination. Specific ubiquitination sites in H2B have been shown to be critical for mitotic and meiotic cell cycle progression (21). Monoubiquitination of H1 contributes to activation of eukaryotic transcription (22). The many histone tail modifications function in concert with each other in order to regulate the chromatin structure. The modifications may also serve as a marker for the recruitment of other regulatory proteins. Chromatin remodeling complexes Although modification of the outside histone tails regulates interactions, it is not likely that it will disrupt the nucleosome core. Alteration of the core structure is accomplished by chromatin remodeling complexes (23-25). Using energy from ATP hydrolysis, these complexes are capable of either removing histones or changing the path of DNA around the nucleosome. Genetic studies revealed two classes of remodeling complexes, the SWI/SNF (named after the mating type SWItching and Sucrose Non-Fermenting yeast genes) and ISWI (named after a drosophila ATP hydrolyzing protein called Imitation SWItch) (25). Both classes include homologous complexes from yeast, drosophila and humans. The SWI/SNF family of remodeling complexes destabilizes the nucleosome by disruption of the histone-DNA contacts in order to make the DNA accessible. A yeast complex of this family has been found capable to transfer an entire octamer of histone proteins to another DNA region (26). A human SWI/SNF homologue is found in a deacetylase complex that disrupts the nucleosome and facilitates deacetylation of the histone tails (24). The ISWI family of remodeling complexes does not disrupt the nucleosome, but enables sliding of histone octamers to adjacent positions in order to create nucleosome free regions. These complexes primarily interact with the histones in order to create a more mobile nucleosome. Two drosophila complexes of this ISWI family have shown to be able to reposition the intact histone octamer along a stretch of DNA (27, 28). While the ATP-dependent remodeling factors can facilitate histone modifications, they can also be regulated in some circumstances by histone modifications. The concerted action of histone modifiers and chromatin remodeling complexes may facilitate the activation and repression of transcription. RNA Polymerase II transcription machinery Transcription of eukaryotic class II genes is a complex biochemical process that is subject to chromatin repression. The essential DNA-binding sites are blocked in the nucleosome itself or made inaccessible by higher-order folds of the (hetero)chromatin structure (6). Chromatin remodeling and histone modifying activities, allows DNA-binding of proteins that are involved in transcription. In general, activation of transcription starts with binding of activator proteins to DNA sites that are adjacent to the start site of transcription (Fig. 1). The activator proteins are involved in the recruitment of chromatin modifying and remodeling enzymes (29, 30) and in the assembling of the general transcription factors (GTFs) to the promoter region (31). Once the TATA-box promoter region is made accessible, the general transcription factors including TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH and RNA Polymerase II (RNAPII) can be assembled in order to form a pre-initiation complex (PIC) (32, 33). A schematic overview of the PIC is shown in Figure 3. Whereas the common set of transcription factors is sufficient for transcription, additional cofactors are required for the response to activators (34-37). The assembly of the PIC starts when the TATA-binding protein (TBP) subunit of TFIID recognizes and binds the core promoter region. The assembly of the PIC is either completed by the sequential association of the basal transcription factors or by recruitment of a pre-assembled TFIID-deficient holoenzyme complex (38, 39). The stepwise assembly model is frequently questioned, as it appears to be rather inefficient to assemble the large PIC within the time scale of the transcription process. The universal pre-assembled RNAPII complex could facilitate a rapid response, but it does not fit well with the vast diversity of cofactors and the distinctly different elongation complex, which would suggest some kind of recycling of shed components. PIC formation is therefore more likely to be facilitated by recruitment of (sub)complexes in which the composition may vary for different promoters. Alternatively, it has been suggested that the nucleus contains a network of transcription active compartments (40). After initial remodeling events, the promoter is recognized within a transcription initiation compartment. Upon deactivation of signals, the promoter can be released and translocated to an adjacent elongation compartment to facilitate RNA processing. 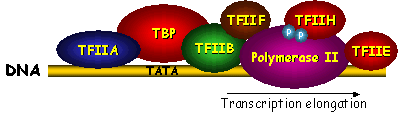 Figure 3. Schematic overview of the pre-initiation complex (PIC). The basal transcription factors are assembled around a TATA-box promoter. Phosphorylation of a subunit of RNA Polymerase II is thought to be critical in the onset to transcriptional elongation (75). TFIID binding to the core promoter The central basal transcription factor TFIID, consists of TBP and a number of TBP-associated factors (TAFIIs). TBP is essential for transcription of class I, II and III genes. Binding of the saddle-shaped TBP through minor groove contacts with the core promoter, results in a sharp bend in the DNA (41). Deformation of DNA by TBP can produce a more compact and stable complex and coordinates the PIC assembly by bringing remotely bound transcriptional activators nearer. TFIID precludes packaging of the core promoter with the histone octamer and provides a platform for further PIC assembly. The TAFIIs are usually required for the activator dependent recruitment and stabilization of TFIID at the core promoter. It is suggested that the TAFIIs may function as coactivators to participate in activation of transcription (42-44). The TAFIIs are present in various compositions in TFIID and some of them have enzymatic activities. Analysis of the molecular organization of TAFIIs revealed that the histone fold is also a structural motif of TFIID (45). Surface expansion and TBP stabilization by TFIIA and TFIIB The transcription factor TFIIA is frequently not regarded as a general transcription factor and resembles the TAFIIs as it is required for activator-dependent transcription, but dispensable for basal transcription on most promoters (46). Characterization and cloning of the TFIIA subunits suggests that it could be best regarded as a cofactor (47). TFIID and TFIIA assemble on the promoter independently of the holoenzyme complex (48). TFIIA consists of three subunits (a, b and g) and stabilizes the TFIID or TBP binding to the core promoter. The crystal structure of the yeast TFIIA-TBP-TATA complex shows that TFIIA is composed of a beta-barrel and a bundle of four a-helices (49, 50). The beta-barrel domain binds the amino-terminal side of TBP and the DNA major groove upstreams of the TATA box. The bundle of a-helices points away from the TBP-TATA complex and offers a substantial surface for interaction with additional transcription factors. TFIIB is one of the essential transcription factors that enters the PIC subsequent to formation of the TBP-DNA complex. This single polypeptide includes an amino-terminal zinc binding domain (TFIIBn), two structurally similar repeats in the carboxy-terminal core domain (TFIIBc) and a phylogenetically conserved sequence that links the two domains. The TFIIBn zinc-ribbon structure has been solved by NMR and shows resemblance to the elongation factor TFIIS (51, 52). The crystal structure of TFIIBc bound to the TBP-TATA complex shows distinct contacts of TFIIBc to the carboxy-terminal end of TBP and to specific DNA bases in the major groove upstream and in the minor groove downstream of the TATA-box (53, 54). The NMR solution structure of the free form TFIIBc shows clear differences with the TBP-TATA bound crystal structure. This indicates that TFIIB undergoes a conformational change upon binding to the TBP-TATA complex (55). The TFIIBc conformation is also affected by interaction with either TFIIBn or the VP16 activation domain (56). It is suggested that TFIIB forms a downstream surface that could act as a bridge between TBP and the RNAPII transcription start site. The structures of the TBP-TATA complexes with TFIIA and TFIIB can be combined to create a model for the TFIIA-TFIIB-TBP-TATA quaternary complex (Fig. 4). This model shows that both transcription factors cooperate to stabilize TBP binding to DNA (41). The surface of the TFIIA-TFIIB-TBP platform on the core promoter can interact with TAFIIs, initiation factors, activators and coactivators. 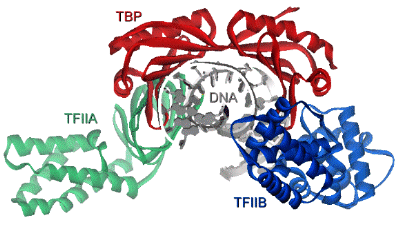 Figure 4. Model of the TFIIA-TFIIBc-TBP-DNA quaternary complex, based on the crystal structures of TFIIA-TBP-TATA (50) and TFIIBc-TBP-TATA (53). View from a position downstream of the TATA box. TFIIF and TFIIE wrap DNA around RNA Polymerase II The transcription factor TFIIF is physically associated with RNA Polymerase II and essential for initiation and elongation. TFIIF interacts with TFIIB, TAFII250 and DNA, and is required for the recruitment of RNAPII into the PIC (57-59). TFIIF is a heterotetramer composed of two heterodimeric subunits composed of the RNA polymerase-associated proteins RAP30 and RAP74 (also referred to as TFIIFb and TFIIFa, respectively). The crystal structure of the amino-terminal parts of the RAP30-RAP74 dimer (Fig. 5) revealed a triple b-barrel dimerization fold (59). The NMR solution and X-ray crystal structures of the carboxy-terminal domains of RAP74 (60, 61) and RAP30 (62) (Fig. 5) show winged helix structures, that resemble the winged helix of the linker histone H5. Similar to the linker histone, RAP30 binds preferably to bent DNA, which could explain the requirement of TBP induced bending of the core promoter. The surface electrostatic properties of RAP74 are essentially different from RAP30, which argues for different functional properties. The RAP74 winged helix has been shown to directly interact with the carboxy-terminal domain of the TFIIF-associated CTD phosphatase (FCP1) (61, 63). Similar to TFIIF, TFIIE is a heterotetramer transcription factor composed of two TFIIEa and two TFIIEb subunits. TFIIE is involved in promoter opening and in the recruitment and regulation of TFIIH (64). The TFIIEb subunit binds to the region where the promoter starts to open. The NMR solution structure of the TFIIEb core domain (Fig. 5) shows another winged helix motif and a DNA-binding surface for opened DNA (65), hereafter referred to as bubble DNA. TFIIF and TFIIE are cooperatively involved in isomerization of the PIC by tight wrapping of the promoter DNA around RNA Polymerase II (66, 67). 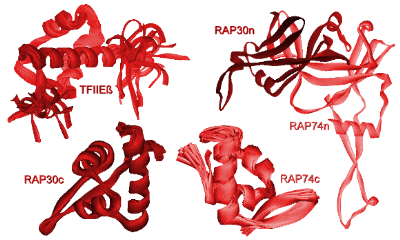 Figure 5. Winged helix structures of TFIIE and TFIIF. NMR solution structures of the TFIIEb core domain (65) and the carboxy-terminal domains of RAP30 (62) and RAP74 (61). The tripartate b-barrel fold of the amino-terminal parts of the RAP30-RAP74 crystal structure is shown on the right side (59). TFIIH facilitates promotor opening The transcription factors TFIIE, TFIIF and TFIIH facilitate together the early events for the transition to the elongation phase of transcription (68, 69). The general transcription factor TFIIH is a large multiprotein assembly that consists of nine subunits. TFIIH has well defined enzymatic activities and is involved in promoter opening and escape, DNA damage repair and cell cycle regulation (70, 71). Among the catalytic activities that TFIIH supports are two ATP-dependent helicases ERCC2 and ERCC3 (also referred to as XPD and XPB, respectively) that unwind the promoter around the start site of transcription in opposite directions. ERCC3 exhibits a 3'-5' DNA helicase activity and facilitates in the downstreams extension of the transcription bubble DNA. The helicase activities are also involved in the DNA excision repair machinery (72, 73). Furthermore, TFIIH contains a cyclin-dependent protein kinase (cdk7) activity which can phosphorylate the carboxy-terminal domain (CTD) of the largest subunit of RNA Polymerase II. This event requires ATP and is mediated by the ERCC3 helicase. Phosphorylation of the CTD is important in promoter clearance and switching RNAPII from the initiation to the elongation phase of transcription (74, 75). The cdk7, cyclin H and MAT1 subunits of TFIIH are part of the cdk-activating kinase (CAK). The CAK is known to be regulated by interaction with specific inhibitor molecules and by (de)phosphorylation of the cyclin H component by either cdk8 / cyclin C (also referred to as SRB10/11 in the Mediator complex) or the casein kinase II (CKII) (76, 77). ERCC3 and four associated proteins (p62, p52, p44 and p34) form the core of TFIIH. The remaining ERCC2 helicase can be found associated with either the TFIIH-core or the CAK and is believed to bridge both activities (71). ERCC3 contacts DNA on both sides of the transcription start site to catalyze promoter melting (78). The upstream contacts require tight wrapping of DNA around RNAPII as is stimulated by TFIIE and TFIIH (Fig. 6). TFIIH is proposed to function as a molecular wrench that rotates downstream DNA to facilitate the unwinding of DNA at the start site of transcription (79). A three dimensional electron microscopy structure of the ensemble of yeast TFIIH subunits shows a ring shaped globule that could accommodate double stranded DNA inside (80, 81). The solution structure of the p44 subunit, that has a role in promoter escape, has been elucidated by NMR and reveals close homology with the regulatory domain of the protein kinase C (PKC) (82). The amino-terminal part of p44 plays an important role in the regulation of the ERCC2 helicase activity. The carboxy-terminal domain is essential for TFIIH transcription activity and binds three zinc atoms. The solution NMR structure of the MAT1 subunit of CAK exhibits a highly positively charged ring finger domain with a conserved hydrophobic central b-strand and an additional a-helical segment in the amino-terminal part (83). The MAT1 ring finger domain might interact with other factors of the basal transcription machinery and allows optimal phosphorylation of the RNA polymerase II CTD.
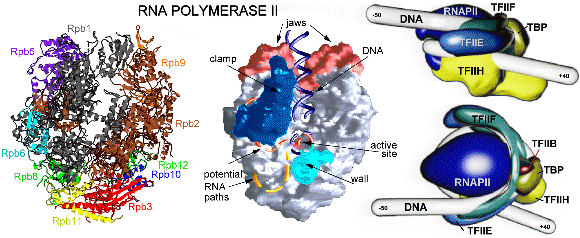
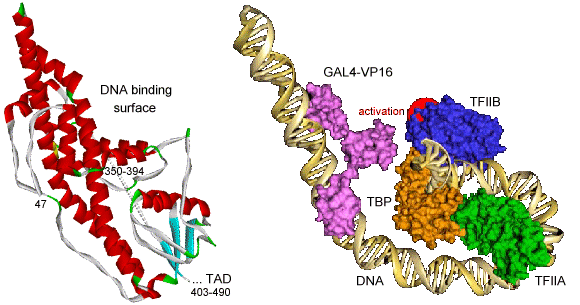
Elongation of RNA Polymerase II through chromatin The engine of transcription, RNA Polymerase II, is a large enzyme that is composed of at least 12 subunits and is remarkably conserved among species. RNAPII is capable of DNA unwinding, RNA polymerization and proofreading (84). The largest RNAPII subunit (Rpb1) comprises a carboxy-terminal domain (CTD) that can be phosphorylated and plays a key role in the transition from transcription initiation to elongation (75). Phosphorylation of the CTD is accomplished by at least four cyclin-dependent kinases (Kin28, Srb10, Ctk1 and Bur1) which have different functions and may regulate transcription at different stages during the cycle (85). The crystal structure of yeast RNA Polymerase comprising 10 protein subunits was elucidated at 2.8 Å and 3.1 Å resolution and revealed a division in four mobile modules (86). Another crystal structure was elucidated at 3.3 Å by molecular replacement of the 2.8 Å structure and shows the RNAPII in the act of transcription (87). The core of the enzyme is formed by the largest subunits Rpb1 and Rpb2 and is surrounded by the remaining subunits (Fig. 6). The catalytic site that contains magnesium ions is formed by a deep cleft in the interface of Rpb1 and Rpb2 and could accommodate about 20 DNA base pairs. The ribonucleoside triphosphates are proposed to gain access to the active site through a pore beneath the catalytic center. The structures reveal the enzyme in an open and partially closed state that mainly differ in the position of the so called clamp region. This clamp consists for the greater part of the Rpb1 subunit and smaller portions of Rpb2 and Rpb6 and allows entry of DNA into the positively charged active center and is thought to fold over the DNA as it enters the enzyme. The position of the clamp may be regulated by phosphorylation of Rpb6 (88, 89). The RNAPII elongation structure clearly shows the synthesis of RNA and helps to understand the structural basis of transcription at the molecular level (69, 90). Newly synthesized RNA exits the enzyme through a groove that ends near the CTD, which is consistent with its key function in transcription. Recently a crystal structure of the complete 12 subunit RNA Polymerase II was solved at 4.2 Å resolution, showing the general location of the missing Rpb4 and Rpb7 subunits (91). A previously reported Rpb4-Rpb7 complex from Archaea revealed a role in ssRNA binding, which is consistent with its position near the exit of synthesized RNA (92, 93). The elongation of RNAPII is accompanied by a number of elongation factors, that interact directly with the enzyme (68). Several factors have been shown to facilitate RNA Polymerase II elongation through the chromatin of the entire gene. Possibly disruption of the nucleosome-DNA binding enables traveling of RNAPII around the nucleosome units. (94). Transcriptional activators Transcription requires dynamical cooperation of many proteins, which are frequently associated in multisubunit complexes (40). The action of the RNA Polymerase II transcription machinery is controlled by specific regulators. It is commonly believed that the proteins involved, such as chromatin remodeling enzymes, transcription factors and polymerase, are stepwise recruited in preassembled (holoenzyme) complexes, in which the actual order and composition may vary for the different promoters (38, 39, 95). The transcriptional activators enhance the basal level of transcription by recruitment of these subunits and complexes, either to modify the chromatin structure (29, 30) or assemble the GTFs on the promoter region (31). The activator proteins are composed of a DNA-binding domain that specifically binds DNA upstream or downstream of the core promoter. In addition, they contain an important transcription activation domain (TAD) that is involved in protein-protein interactions (96). These TADs are commonly classified on the basis of their most abundant amino acid content, which results in different groups that are rich in glutamine, proline, serine/threonine or acidic residues (96, 97). Despite this classification, specific hydrophobic and aromatic residues are often determined to be more critical for the activation function than these most abundant residues (98-104). TADs often lack a folded structure, but display a highly flexible random-coil-like conformation under physiological conditions (96, 105, 106). Typically, the intrinsically unstructured TADs undergo induced folding when binding to their targets (106). The induced formation of an a-helical structure was shown for TADs containing an FXXFF motif (F: hydrophobic residue, X: any amino acids). These general short recognition motifs fold into amphipathic a-helical regions to create a hydrophobic interaction surface on one side of the a-helix. Examples are VP16 (107), p53 (108, 109) and CREB (110, 111). In contrast to this induced a-helix formation, the E2 activation domain of the papillomavirus (112) and the TAD of the activating transcription factor ATF-2 (113) are already (partially) structured in the free form. The yeast heat shock transcription factor remains unstructured, even in presence of its DNA target (114). The tumor suppressor p53 has been shown to exhibit a-helical propensity in isolation (115) and induce structure upon binding to hTAFII31(109) and the MDM2 oncoprotein (108), although it binds in an unfolded form to the chaperone Hsp90 (116). Detailed studies of the kinase-inducible activation domain (KID) of CREB reveals induced folding of two orthogonal a-helices upon binding to the KIX domain of CBP, but only when it is induced by phosphorylation at S133 of KID (110). The activation domain of the proto-oncogene c-Myb however, binds to the same site on the KIX domain, without the requirement of phosphorylation. This TAD spontaneously adopts a-helical structure, even as an isolated peptide (117). Comparative binding studies of c-Myb and KID domains show that binding of KID is driven by enthalpy while c-Myb is entropically favored, suggesting different mechanisms for activation. It is suggested that TAD binding is a multistep process (118), in which a low affinity complex is formed by initial binding (driven by electrostatic interactions). This unstable complex slowly converts to a more defined and stable form by specific (hydrophobic) contacts between the TAD and its target. The herpes simplex virion protein 16 (VP16) Model systems as the Herpes Simplex Virus (HSV) have been investigated thoroughly to understand the complex regulation of eukaryotic gene expression (119). When HSV infects a cell it can enter a latent state and reside for a long period in the host cell, or it can enter a lytic program in which the infected host cell is finally killed (120). The two modes of infection involve intimate interactions between the virus and the host cell. HSV is not dependent on cellular S-phase induction since it encodes its own DNA replication machinery. Three groups of virus-specific proteins, designated as immediate early (IE), early and late, are synthesized in a coordinated and sequential fashion during infection. The IE gene expression has been studied extensively as the encoded proteins play an essential role in the regulation of the viral gene expression. The herpes simplex virion protein 16 (VP16) is a key player in the choice between lytic and latent infection as it is important for the initiation of the lytic program through control of the IE gene expression. The VP16 protein was first identified in 1984 as a trans-acting polypeptide sequence responsible for stimulation of IE gene transcription (121). VP16 has been designated before as a gene trans-inducing factor (a-TIF), infected cell protein 25 (ICP25) or virion molecular weight 65 kDa (VMW65). The VP16 activator forms a complex with the cellular proteins and mediates IE gene expression through a conserved upstream regulatory element, designated as TAATGARAT. The VP16 protein first associates with the POU domain octamer-motif binding factor Oct-1 and the host cell-proliferation factor HCF-1 to bind to the regulatory promoter element. IE gene transcription is subsequently activated by the very potent VP16 transcriptional activator. VP16 itself is a product of late-gene expression and incorporated into the newly synthesized virions for the next round of infection. Biochemical and molecular genetic methods showed that the VP16 protein can be divided into two domains (122, 123). The 490 amino acid polypeptide contains a core region and a carboxy-terminal region (residues 410-490), enriched in acidic residues, which acts as a TAD (122, 123). The largest part, the amino-terminal domain, directs VP16-induced complex formation, contains a DNA-binding surface and is conserved among many different herpesviruses, including the human varicella-zoster virus (VZV) causing chicken pox and shingles, the chicken Marek's disease virus (MDV), the equine herpesvirus (EHV-1) and the bovine herpesvirus (BHV-1). The mostly a-helical structure of the 47-402 core region has been elucidated by crystallography at 2.1 Å resolution, except for 45 residues (350-394) that lacked sufficient density (124). This core region possesses a seat like structure (Fig. 7) which binds the specific DNA region. The unstructured region enhances the DNA binding affinity of VP16 and interacts with Oct-1, HCF-1 and DNA, suggesting a bidentate structure that contacts the TAATGARAT element (124, 125). In addition, HCF-1 contains a transcription activation domain, which cooperates with the TAD of VP16 to activate transcription (126).
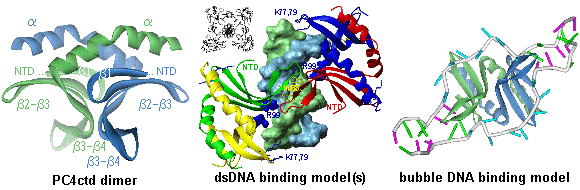
The VP16 activation domain (VP16ad) enhances transcription rates by promoting the assembly of the PIC through interactions with GTFs or recruitment of the holoenzyme complex (127) and by stimulating the formation of an open promoter complex (128). VP16ad targets many factors of the RNA polymerase II transcription machinery, like TBP (129), TFIIA (130, 131), TFIIB (132), the RAP74 subunit of TFIIF (133), the p62 component of TFIIH (134), the TBP-associated-factors hTAFII31 (107), dTAFII40 (135), hTAFII32 (136), the Mediator complex (137-140) and the cofactors PC4 (141, 142), CBP (143, 144) and p300 (145, 146). VP16ad stimulates assembly of the PIC by increasing the amount of correctly oriented TBP bound to the TATA box (147). In addition, VP16 competes with dTAFII230 for binding to TBP (148), which might alleviate the inhibition of TBP-TATA binding by this TAF. Furthermore, it has been shown that VP16 can modify the cell-cycle dependent change in chromosome position and is involved in chromatin unfolding and remodeling (149, 150). VP16ad interacts with many components of the chromatin remodeling complexes (SWI/SNF) and histone tail modifiers (HAT) (151, 152). The VP16 activator can sequentially recruit these histone tail modifiers and remodeling enzymes, suggesting that VP16 can activate the target genes within the context of the condensed chromatin (153). VP16ad can be subdivided into two functional regions, each of which is independently capable to activate transcription in vivo via distinct pathways (99, 122, 135, 144, 154, 155). A minimal pentapeptide motif was found to be sufficient for strong transcriptional activation (156). Extensive mutational analysis revealed key roles for specific hydrophobic residues in both subdomains (98, 99, 155, 157). When the critical F442 residue is mutated to proline, the interaction with TFIIB and TBP are disrupted in vitro (132, 158, 159) and binding to TFIIH (134) and PC4 (142) is substantially reduced. Structural studies of VP16ad showed that the TAD is unstructured in its free form (107, 160, 161). However, structure may be induced when binding to specific target proteins (107, 162). Based on site-specific protein-DNA photo-cross-linking experiments, a model is proposed for the (direct) interactions between the VP16 activation domain (fused to the DNA binding domain of the yeast activator GAL4) and the TBP-TFIIA-TFIIB-DNA quaternary complex (Fig. 7) (163). Transcriptional cofactors Transcriptional cofactors are intimately associated with components of the basal transcription machinery and can be involved in promoter recognition and enzymatic functions. These proteins are frequently also called coactivators as they are recruited to the promoter by DNA-bound activators to mediate interactions that enhance or repress transcription. Many cofactors contain enzymatic activities that can either target the chromatin or the basal machinery to influence transcription. For example, the CREB binding protein (CBP) contains a HAT activity, which can target histones and transcription factors as p53 and Pit-1 (164, 165). Similarly, the HAT activity of the p300 cofactor, directly involved in chromatin-mediated transcription activation, can also acetylate the basal transcription factors TFIIEb and TFIIF and other cofactors as p53 and PC4 (165-167). Interestingly, acetylation of both cofactors is inhibited by phosphorylation. Furthermore, the silencing mediator for retinoid and thyroid receptors (SMRT) and the nuclear receptor core-repressor (NCoR) interact with basal transcription factors and form complexes with HDAC, which targets the chromatin (168-170). In general, the cofactors are subdivided in three distinct classes: TAFIIs, the Mediator and general cofactors (171). As discussed above, the TBP associated factors and TFIIA can function as coactivators that mediate the interactions towards activators and core promoter elements. The Mediator is important for activation and repression of transcription and plays an essential role by direct contacts with RNA Polymerase II and modulating the activity of the enhancers and operators (35, 36, 172). The Mediator comprises about 20 subunits and is known to be present under many different names and compositions for yeast, mouse and human complexes (such as TRAP/SMCC, NAT, DRIP, ARC and CRSP) (139, 173). Some of the proteins are conserved between species. Many proteins found in the Mediator complex overlap with the RNAPII holoenzyme and have multiple names. The variation in Mediator composition might be caused by the process of purification and identification, but may also be specifically related to the various promoters (173, 174). Some subunits of the Mediator complex have been found to directly interact with the CTD of RNA Polymerase II or regulate the TFIIH CTD kinase activity. Low resolution (30-35 Å) electron microscopy structures of Mediator complexes show an extended oval structure that evolves in presence of RNA Polymerase II to a crescent shaped structure with a head, middle and tail domain (175, 176). The head is highly conserved among yeast, mouse and human Mediator complexes, and makes extensive contacts to RNAPII. The tail domain is the least conserved part and exhibits various shapes among the different Mediator complexes. Many general cofactors were originally isolated from upstream-factor stimulatory activity (USA) that augments activator-dependent transcription of class II genes (177). Several coactivators in this fraction were characterized and termed positive cofactor (PC) whereas the repressors were named negative cofactor (NC) (171). The negative cofactor NC2 was later identified to be the high mobility group protein HMG1 (178). The cofactors PC1 and PC3 turned out to be identical to poly ADP-ribose polymerase and DNA topoisomerase I, respectively (171, 179). Furthermore, PC2 contains a subset of proteins present in the Mediator complex (174). The positive cofactors PC3 and PC4, have been shown to enhance and repress transcription under specific conditions (179-181), showing that the distinction between PCs and NCs is not unambiguous. The structure of the NC2-TBP-DNA ternary complex (182) provides more insight in the mechanism of transcriptional repression by NC2. The two NC2 subunits, NC2a and NC2b resemble the core histones H2A and H2B, respectively. Superposition of this structure with the TFIIB-TBP-DNA and TFIIA-TBP-DNA ternary complexes suggests that NC2 represses transcription by blocking the recruitment of TFIIA and TFIIB to the preformed TBP-DNA complex. Like the TADs of activators, some cofactors also possess intrinsically unstructured domains. The steroid receptor co-activating factor-1 (SRC-1) and the glucocorticoid receptor interacting protein 1 (GRIP-1) are examples of cofactors that contain the typical FXXFF motif (as discussed before for the TADs) and show induced formation of an amphipathic a-helical structure upon complex formation (183-185). The amino-terminal domain of drosophila TAFII230 is unfolded in solution and induces structure when it interacts with TBP (186). This domain folds into three a-helices and a small b-sheet to mimic the minor groove hydrophobic and charged surfaces of the TATA element that matches the saddle shaped surface of TBP. In this way it negatively regulates the DNA-binding activity of TBP within the TFIID complex. Furthermore, the B cell-specific transcription co-activator, Bob-1 (also known as OCA-B or OBF-1) has little secondary structure, but may become partially structured by binding to the DNA-bound POU domain of Oct-1 (187). Many more proteins involved in the regulation of the transcription process contain these intrinsically unstructured domains, which are easy targets for modification (like histone tails) and may be involved in autoregulation and binding to their targets (105, 106, 188, 189). The general transcriptional cofactor PC4 Molecular cloning and characterization of embryonal tissue and chemically induced cancers from rats led in 1984 to the discovery of a highly transcribed gene, expressing a specific protein, later known to be a homologue of PC4 (190). A single-stranded DNA (ssDNA) binding protein was purified from murine plasmacytoma nuclear extracts in 1988 and revealed to be a proteolytic fragment of a larger polypeptide (191). It was strongly suggested that this mouse protein, corresponding to PC4, was evolutionarily conserved in both mouse and human tissues. Both the groups of Meisterernst and Roeder cloned and characterized the full length PC4 protein in 1994, through purification of a protein fraction from HeLa cell nuclear extracts that stimulated transcription (141, 142). Deletion analysis of the 126 residues protein revealed a bipartite structure of PC4. The carboxy-terminal half comprises a ssDNA binding domain (PC4ctd, residues 61-126). Interestingly, full length PC4 interacts weaker with ssDNA than PC4ctd, but is on the other hand capable of low affinity double-stranded DNA (dsDNA) binding, which has been shown to correlate with the ability of PC4 to mediate activator dependent transcription (141, 192). The amino-terminal domain (PC4ntd, residues 1-60) has a remarkable amino acid composition, consisting of a lysine rich region in between two serine rich regions. The first serine rich region also contains many acidic residues and is therefore referred to as serine/acidic (SEAC) rich region. PC4 interacted independently with free and DNA-bound VP16 activation domains, free and DNA bound TFIIA-TBP complexes, but not with TBP alone or in complex with TFIIB. Surprisingly, the yeast homologue Sub1/Tsp1 of PC4 fails to bind TFIIA, but specifically inhibits the TBP-TFIIB complex formation by interaction with TFIIB (193, 194). This yeast homologue of PC4 increases the off-rate of TFIIB and thereby reduces the frequency by which transcription is initiated. PC4 was shown to facilitate activation in response to many activators as VP16, AH, CTF, SP1, E1a, IE and NF-kB (141, 142). Since these activators represent all major classes of transactivation domains (acidic, proline rich or glutamine rich), PC4 is referred to as a general or promiscuous cofactor. Since PC4 does not only bind to activators and general transcription factors, but also to ssDNA and dsDNA it is an exceptionally versatile cofactor. The structure of PC4ctd has been elucidated by crystallography at 1.74 Å (Fig. 8) and reveals a homodimer with two symmetrical b-channels running in opposite direction (195). The monomer consists of a curved four-stranded anti-parallel b-sheet followed by a 45� kinked a-helix. The hydrophobic surface of the a-helix of one monomer and the b-sheet region of the other monomer forms the dimer interface. In addition, the crystal structure shows multimerization between the PC4ctd dimers by either the a-helix regions or the b2-b3 loop regions. The buried surface area of these regions is only about 580 Å2 or 1000 Å2 respectively, compared to ~3000 Å2 for the dimer interface. The tetrameric arrangement of PC4ctd dimers demonstrates a spiral axis with a central positive surface. Although PC4ctd is unable to bind dsDNA, the spiral diameter and position of arginine 99 appear suited to wrap around the dsDNA phosphate backbone (Fig. 8). This hypothetical dsDNA binding mode, most probably requires further stabilization by PC4ntd. The ssDNA binding surface of PC4ctd has been identified by NMR (196). Amide resonance shifts were observed for charged and hydrophobic residues at the two b-channels upon binding the oligonucleotide dT18. The b-channels and loops show above average flexibility in solution, which is reduced in the presence of ssDNA. On the basis of the channel length of nearly 40 Å and similar binding as observed for the replication protein A (RPA) to ssDNA (197), 8 nucleotides are expected to bind. It is very likely that the role of PC4 in ssDNA binding involves binding to opposing strands of destabilized dsDNA, which results in a juxtaposed arrangement of single strands (198). A putative binding model (Fig. 8) illustrates how PC4ctd can interact with a DNA bubble.
PC4 forms complexes with the trimeric RPA protein on ssDNA, which influences the DNA replication function of RPA, suggesting a possible role of PC4 in the initiation of DNA replication (199). Comparable to PC4, RPA also binds ssDNA and is composed of a global b-barrel fold and an unstructured flexible tail (200). It was recently shown that phosphorylation of the tail induces intersubunit interactions between the negatively charged tail and a basic cleft of the DNA-binding domain, which modulates the interactions of RPA with dsDNA (201). Similarly to RPA and histone tails (5-7), the amino-terminal tail of PC4 enables regulation, interactions and multimerization. Likewise, the SEAC region of the amino-terminal domain can be phosphorylated, which negatively regulates the cofactor function but not the ssDNA binding activity of PC4 (192, 202). Since 95% of PC4 is predominantly present in this (inactive) phosphorylated form in vivo (202), phosphorylation could be the crucial step in regulation of the cofactor function. The lysine rich region of the PC4ntd tail is involved in binding to dsDNA (192). The binding affinity of PC4 for dsDNA is increased by 40% when two lysine residues are acetylated by the cofactor p300 (167). It has been proposed that the loss of dsDNA binding upon phosphorylation can be caused by masking of the functional lysine rich region by the phosphorylated SEAC motif (192), a model that could also explain the inability to acetylate phosphorylated PC4 (167). The lysine rich region is also shown to be important for the ability of PC4 to inhibit cdk1, cdk2 and cdk7 mediated phosphorylation of RNAPII (203), an event thought to be critical in the onset to transcriptional elongation (75). The phosphorylated form of PC4 is not able to inhibit the phosphorylation of RNAPII, which suggests a role for PC4 in the conversion to the elongation phase of transcription. Both PC4ctd and full length PC4 destabilize dsDNA by local strand separation (198). This unwinding activity suggests a possible involvement of PC4 in promoter opening and other strand displacement events. Similar ATP-independent unwinding activities have been observed for the bacteriophage F29 protein P5 (204) and the adenovirus DNA-binding protein (DBP) (205). Contrary to transcriptional stimulation, PC4 has been shown to repress transcription in the absence of TAFIIs and TFIIH (180) or in the absence of an activator (206). This inhibitory activity of PC4 has been correlated with binding to a DNA bubble (207). The combined effects of TAFIIs, TFIIH and a preassembled RNA Polymerase II holoenzyme have been show to attenuate repression of transcription (180, 206-209). Recently, it has been shown that repression is alleviated by the ERCC3 helicase activity, present in TFIIH (210). Accordingly, the foremost function of PC4 could actually be to repress transcription until PIC assembly is complete, as TFIIH is generally believed to be the last factor to enter the PIC (33). PC4 maps to the human chromosome 5p13. This location is frequently associated with loss of heterozygosity in lung and bladder tumors (211, 212). This suggests that the transcriptional cofactor PC4 may have a role in tumor suppression. Indeed, PC4 has been reported to interact with the proline and glutamine rich activation domains of AP-2 (213). Overexpression of PC4 restores the activity of this activator in ras-transformed PA-1 cells. The PC4 overexpressing ras cell lines had a diminished growth rate and exhibited a loss of tumorigenicity in nude mice. PC4 also interacts with the breast and ovarian specific tumor suppressor protein, BRCA1(214). The ability of BRCA1 to activate transcription was largely enhanced in the presence of PC4 and strongly affected by TFIIH concentrations. This suggests again that PC4 regulates transcription in a TFIIH dependent fashion. The cofactor PC4 has been identified to interact with the transactivation response element (TAR) of the human immunodeficiency virus (HIV) transactivator Tat (215). Overexpression of PC4 caused enhanced tat-dependent activation of the HIV long terminal repeat. Both the PC4ctd core and the PC4ntd lysine rich region were shown to be important for the interaction. Phosphorylation of PC4 and mutation of residues in the lysine rich region prevented interaction with Tat. Two other human transcriptional coactivators p52 and p75 resemble some aspects of PC4 as they serve as adaptors between similar sequence specific activators and possibly other components of the basal machinery (216). Interestingly, these coactivators also interact with PC4. An evolutionary conserved link between transcription and polyadenylation of mRNA precursors has been demonstrated by the interaction of PC4 with the polyadenylation factor CstF-64 (217). Both proteins have a yeast homologue (Sub1/Tsp1 and Rna15p, respectively) that have also been shown to interact. It is suggested that PC4 possesses an anti-termination activity. Consistent with this suggestion is the observation that PC4 enhances TFIIIC interactions with downstream promoter regions (218). Since a strong interaction was observed between PC4 and RNAPII, it has been suggested that RNA Polymerase II recruitment can be mediated through concerted interactions of PC4 and the basal transcription machinery (180, 219). Interestingly, a human Mediator complex has been shown to efficiently phosphorylate PC4 as well as the CTD of RNA Polymerase II (219). In another RNAPII containing Mediator complex, PC4 has been shown to be essential to enhance transcriptional activation by GAL4-VP16 (140). Further studies demonstrated the importance of PC4 for transcriptional (co)activation by NF-Y (220), the thyroid hormone receptor (221), the glutamine-rich POU homeo domain protein Oct-1 (222), some of the adeno-associated virus Rep proteins (223, 224), the human papillomavirus E2 (225) and the hepatocyte nuclear factor HNF4a (226). In conclusion, PC4 is involved in many aspects of the transcription machinery as it interacts with activators, recruits GTFs, stabilizes the PIC, binds dsDNA and ssDNA, unwinds dsDNA, interacts with RNAPII and regulates phosphorylation of the CTD. PC4 is furthermore suggested to be involved in tumor suppression, elongation and polyadenylation. Detailed structural and functional studies have to be performed to unravel the working mechanism of the exceptional versatile cofactor PC4 and its involvement within the transcription cycle. Outline of the thesis This thesis describes structural and functional studies on the transcriptional cofactor PC4 and the TAD of the transcriptional activator VP16. The interaction between both transcription factors has been studied as well as the influence of phosphorylation of PC4. In addition, interactions with DNA and some other transcription factors have been investigated. Understanding of the structural and biofunctional aspects of these proteins provides fundamental insights in the molecular biology of gene expression. The specific regulatory role of the amino-terminal domain of PC4 on the cofactor function and ssDNA binding activity of PC4ctd has been investigated and described in Chapter 2. The structural and functional properties of PC4ntd are studied and the PC4 interaction site for the VP16 activation domain is determined. PC4ntd exhibits some tendency to form secondary structure elements and is shown to be highly flexible, even in complex with VP16. PC4ntd is supposed to regulate the PC4 cofactor function through specific interactions with activators and dsDNA and by modulation and/or shielding of the functional surface in PC4ctd. Chapter 3 deals with the crucial role of phosphorylation on the unwinding characteristics of PC4 and the interaction to dsDNA, ssDNA and VP16. It is proposed that the ability of PC4 to perform the required activities during the transcription cycle depends on the phosphorylation status. Finally, the structural and functional aspects of the VP16 activation domain are studied by NMR and mutagenesis and described in Chapter 4. The TAD is affected by interaction with PC4 and a docking model is presented for the binding of VP16ad to the PC4ctd core domain. |