
![]()
Dramatic facts in the exciting field
of genetics are opening up another front in the battle against birth defects.
Thalassemia is a genetic blood
disorder that affects person’s
ability to produce hemoglobin. Hemoglobin is the protein in our red blood
cells that carry oxygen and nutrients to all parts of the body. Without
it, our bodies become weak and unable to thrive. In the past, before the
discovery of its worldwide distribution, it was also known as Mediterranean
Anemia. It comes from the Greek word “thalasanemia” which means “anemia
by the sea”. The Thalassemias (2forms) are among the commonest inherited
blood diseases in the world. Over the centuries they must have accounted
for the death of many millions of children. The clinical picture was not
recognized as a specific disease until 1925, and than not in the one area
of the world where the disorder was most common but in the United States
by a pediatrician, Thomas Cooley. Following his report more cases were
found and the condition became known as Cooley’s anemia. It is known that
Thalassemia occurs not only in people from Mediterranean area but also
in the Middle East, the Indian subcontinent, South – East Asia and Africa
among immigrant populations from these areas.
The Thalassemias have a distribution
concomitent with areas where malaria is common. It is thought that possessing
one of the abnormal genes
that gives rise to Thalassemia
gives some protection against malaria. Some doctors believe that this is
because the red cells in the body of a
thalassemic person are somewhat
fragile, so that when the malarial parasite gets inside the red cell breaks
down and the parasite stops growing. In normal people (non-thalassemics),
the parasite would continue to multiply. So those people with minor forms
of Thalassemia appear to have some protection against malaria. In addition
there is much more information available about Thalassemia and its effects
on the body. This means that medicine can offer more effective treatment
than it could in the past. Due to global migration patterns, there has
been an increase in the incidence of Thalassemia in North America in the
last ten years, primarily due to immigration from Southeast Asia. The disorder
is found in 60 countries. Children born to parents who both carry the trait
have a 1 in 4 chance with each pregnancy of inheriting the fatal form of
the disorder. Not long ago, children born with Thalassemia seldom survived
their first decade of life. Recent medical advances have increased life
span.To stay alive, patients must undergo blood transfusions every two
to four weeks. Every night patients must insert a needle to receive painful
infusions of a special drug for up to twelve hours.There is currently no
cure. Bone marrow transplants and gene therapy procedures whilestill experimental,
suggest that a cure may one day be perfected. Even with extensive treatment,
life expectancy is uncertain.
What are the different kinds of Thalassemia
There are three clinically significant
types:
Thalassemia minor, Thalassemia
intermedia, and Thalassemia major.
Thalassemia minor, also called
Thalassemia trait may cause no symptoms, but changes in the blood do occur.
A person with Thalassemia minor generally has no health problems excepting
a possible mild anemia, which cannot be corrected with iron supplements.
Thalassemia intermedia, also called
mild Cooley’s anemia is an intermediate form of the disease. Thalassemia
intermedia is a clinical condition
that varies and must be constantly
evaluated by the hematologist. No two people with Thalassemia intermedia
are the same.
Thalassemia major, also called
Mediterranean anemia or Cooley’s anemia, named after the doctor who first
described it in 1925) is the most
severe form. Thalassemia major
is very serious and requires extensive medical care.
How do people get Thalassemia?
Thalassemia is an inherited disease,
which means it is passed on from parents to their child through their genes.
It is nor infectious and cannot be “caught” like a cold. It will not develop
later in life, nor can a child outgrow it. Both parents must have Thalassemia
trait in order to pass the
disease on to their child, but
it only takes one parent to pass trait on to his/her child. Thalassemia
trait will never develop into disease. Thalassemia trait can be passed
on for many generations without being detected before a child is born with
disease. The probabilities to get Thalassemia exist for each child independently
of what happened with prior children the couple may have had. In other
words, each new child has a one-in-four chance of having severe Thalassemia.
The inheritance of Thalassemia genes is purely a matter of chance and cannot
be altered. For example if a thalassemic married a normal, all the children
will be healthy carriers. They must inherit a Thalassemia gene from their
thalassemic parent, but they must also inherit a normal gene from the normal
parent, so none of them can possibly haveThalassemia major. (Figure1).
Figure 1
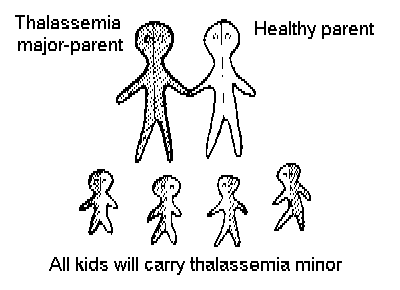
Figure 2 shows that if a thalassemic marries a Thalassemia carrier, in each pregnancy there is a 50% chance that the child will be thalassemic, and a 50% chance that it will be a healthy carrier.
Figure 2
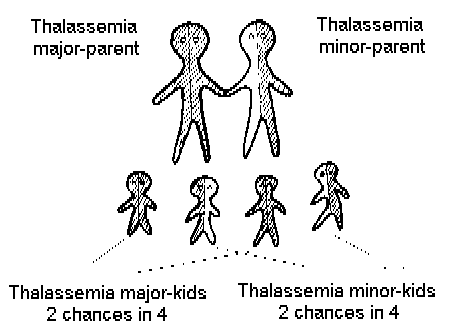
Figure 3 shows that if one thalassemic
marries another, all their children will be thalassemics. This situation
arises sometimes too, because
Thalassemia carriers have so much
in common, that they usually decide not to have any children or to try
to have children with medical help, for example, by artificial insemination
(using semen from a man who is not a carrier).
Figure 3

How long can people with Thalassemia major live?
These days most Thalassemics grow
up to become adults, and earn their own living. Most also find a partner
and get married.Now a number of Thalassemia major patients have their own
children. The disorder and its influence are changing almost from day to
day, because of advances in treatment. Thalassemic patients are now living
longer. Today it is reasonable to think that people with Thalassemia major,
who have been treated from the beginning, may well, live as long as people
without Thalassemia. Only time will tell. Even so, thalassemics live with
more risks than non-thalassemics, because of the amount of medication and
treatment they receive. But all medical treatments include some risk.
Is there a test for Thalassemia?
Yes. Blood tests and family genetic
studies can show whether an individual has Thalassemia or is a carrier.
In addition, parental testing using
chorionic villus sampling (CVS)
or amniocentesis can detect or rule out Thalassemia in the fetus. Early
diagnosis is important so that treatment can prevent as many complications
as possible.
CVS-Chorionic Villus Sample
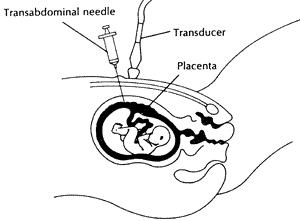
What is Chorionic Villus Sampling
(CVS)?
CVS allows us to take a small sample
of tissue, the chorionic villi, from the developing placenta. CVS is one
of the two tests available to check the chromosomes (the structures that
contain genes) of the baby. This tissue may also be used for biochemical
or direct genetic testing. CVS is performed at 11-12 weeks in the pregnancy
and is a day patient procedure.
An ultrasound will be performed
first to cheek how far pregnant you are, and to show the position of the
baby and the developing placenta. As the baby is small and not fully developed,
very few physical problems can be detected at this stage by ultrasound
scan. The mother's bladder should be full but need not be uncomfortable.
CVS diagram
Chorionic villus can be obtained
by inserting a needle through the mother's abdomen (which has been cleaned
with antiseptic and numbed with
anaesthetic) under ultrasound guidance
into the developing placenta. A small sample of chorionic villi is withdrawn.
Some women experience
discomfort during and after this
procedure and it is advisable to rest for the remainder of the day. The
chorion villus sample is sent to the
laboratory for testing. A chromosome
result will be available in one to two weeks. A biochemical or direct genetic
(DNA) test may take longer. CVS carries a small risk of causing a miscarriage.
The average risk is 1 chance in 100.
AMNIOCENTESIS

What is Amniocentesis (Amnio)?
Amniocentesis is the removal of
a small amount of amniotic fluid from the sac around the baby. This fluid
contains cells which come from the baby and the placenta. This test is
usually performed at 16 weeks in the pregnancy.
Amnio diagram
After cleaning the skin with antiseptic,
a fine needle is inserted under ultrasound guidance through the mother's
abdomen into a pool of amniotic fluid. A small amount of fluid is withdrawn
and the needle removed. Most women say amniocen- tesis is not painful -
however some women feel discomfort. After the procedure it is advisable
to rest for the remainder of the day. You may like to have a family member
or friend with you on the day of the test. The anmiotic fluid is sent to
the laboratory for testing. A chromosome result will take 2-3 weeks. A
biochemical or direct genetic (DNA) test may take longer. Amniocentesis
carries a small risk of causing a miscarriage. The average risk is 1 chance
in 200.
What will happen after the
test?
There is a risk of naturally occurring
miscarriage in all pregnancies. The risk is highest in early pregnancy
and increases with the age of the mother. The risks quoted for prenatal
tests are in addition to this naturally occur- ring risk of miscarriage.Most
people have normal results after the test. However, if the test shows the
baby to have a problem, you will have the opportunity to discuss the results
with your doctor, genetic counsellor or geneticist.
What
stages does a woman with Thalassemia go through before, during
and after pregnancy?
For a woman with Thalassemia to
have children they must have normal sexual development. Many young women
with Thalassemia are not having their periods, or whose periods have started
and then stopped. In this case they can be treated medically so that they
produce eggs. If they are not physically fit, a pregnancy could be risky
for them and the baby. An expected mother should be fit, meaning they must
use their pump regularly, her serum ferritin level should be around 1000
and her heart and liver should not have been damaged by iron overload.
However, even if they are not perfectly fit there is a chance that they
could have a fairly normal pregnancy. Women are advised
to stop Desferal (Desferal is a
medication that removes this excess Iron from the body) when they are trying
to become pregnant, or as soon as they are pregnant. There is no evidence
that Desferal can harm the fetus, but in general it is a good idea for
any pregnant woman to not take
drugs during pregnancy. Mothers
who breastfeed, can start taking Desferal again as soon as the baby is
born. Desferal does not pass from the mother’s body onto milk and so cannot
harm the baby. Before a woman with Thalassemia decides to have a baby they
must take account about the long-term future, their own health, and survival
or whether they will have support from their family’s.
What
is the major treatment now?
The only treatment for Thalassemia
major is regular blood transfusions, usually every three or four weeks.
Most children who have these
transfusions, usually every three
or four weeks, grow normally and live quite happily into their early twenties.
But to live longer, they need other treatments as well. After each blood
transfusion the red cells in the new blood are broken down slowly over
the next four months. The iron from the red blood cells stays in the body.
If this damage is not prevented most people with Thalassemia major die
before they reach twenty years old. At present the only way to remove the
extra iron from the body is to give injections of a drug called Desferal
(Desferrioxamine).

CANE' Srl Via Pavia,
105/i 10090 Rivoli-Cascine Vica (Torino) Italy
Tel.: ++39-011-957.48.72
Fax ++39-011-959.88.80
Internet: www.microjet.it
E-Mail: mailbox@microjet.it
This medication starts between the
ages of 4 to 8 years old. Desferal is injected under the skin. The injections
are given using a portable battery operated pump. The pump has controls
that allow you to set the time for emptying the syringe, and an alarm that
warns you if the syringe gets stuck, or when the drug has all been injected.
The pump is used 5-7 nights of every week. Usually the parents are responsible
for this until the child is able to take over. Desferal picks up the iron
and carries it out in the urine.
This treatment is very successful and most children treated with blood transfusions and Desferal can now lead fairly normal healthy lives. But the treatment is unpleasant and often upsetting, it also interferes with their desire for an active social life and sometimes medication is neglected.
Do Thalassemia person need to be on a special diet?
Thalassemia major patients should
try to keep away from foods high in iron such as red meat, liver, kidney,
green leafy vegetables such as
spinach, some breakfast cereals,
wholemeal breads and alcohol. Although this is recommended, patients do
not have to stick with this diet.
What research in Thalassemia is taking place?
Treatment today is more advanced
then what it was. Scientists are working on better ways to remove excess
iron from the body in order to
prevent or delay iron overload.
They are developing and testing the effectiveness of oral iron-chelating
drugs (L1), which could greatly simplify
treatment of this disease and they
are seeking to develop an effective form of gene therapy that may someday
offer a cure for Thalassemia. The results from the L1 have been good except
of one specific side effect called “Neutropenia” which reduces the ability
of the body to cope with infections so the patient has an increased risk
at infections and can die due to this. Gene therapy may involve inserting
a normal beta globin gene (the gene that is abnormal in this disease) into
the patient’s stem cells the immature bone marrow cells that are the precursors
of all other cells in the blood. Another form of gene therapy may involve
using drugs or other methods to reactivate the patient’s genes for fetal
hemoglobin. All humans produce a fetal form of hemoglobin before birth;
after birth, natural genetic switches “turn off” production of fetal hemoglobin
and “turn on” production of adult hemoglobin. Scientists are seeking ways
to activate these genetic switches so that they can make the blood cells
of patients with Thalassemia produce more fetal hemoglobin to compensate
for their deficiency of adult hemoglobin. Initial studies of rare individuals
with genetic traits that allow them to produce only fetal hemoglobin show
that they are generally healthy, demonstrating that fetal hemoglobin can
be a fine substitute for adult hemoglobin. In addition, improved bone marrow
transplantation methods may lead to wider use of the technique as a treatment
for Thalassemia. Bone marrow transplants have cured some cases of Thalassemia
but they are not widely used.
The disease can’t be prevented at this time, but a program of health education, testing for the trait, genetic counseling, and prenatal diagnosis can provide families with full medical information to help them have healthy children. People who think they may have or carry Thalassemia can go to a genetic services center or clinic for the latest information and for testing. Individuals can be tested to find out if they are carriers.Genetic counselors then can help them make plans about future families.
Why do a thalassemic need genetic counseling?
Genetic counseling is a communication
process of providing information and support to families, couples, or individuals
that are in some way
impacted by an inherited disease
such as Thalassemia. A genetic counselor often acts as a liaison for communicating
complicated medical and
genetic information. A non-directive
approach can help counselors integrate this information into their own
system of beliefs and values. In this
way, counselors can make informed
personal decisions about genetic testing, health care, and reproduction
that make sense for them and their
families.
Iron deficiency or Thalassemia?
Thalassemia and iron metabolism are closely linked. Iron deficiency and mild forms of Thalassemia (E.G. Thalassemia trait) are often confused. Both are associated with mild to moderate anemia and microcystosis (small red cells). At the other end of the spectrum, severe forms of Thalassemia frequently produce iron overload. Excess iron accumulates due to enhanced iron absorption produced by Thalassemia, repeated blood transfusions or both. A number of questions are frequently asked regarding Thalassemia and iron.
Should
a person with Thalassemia trait avoid iron, such as iron-fortified
vitamins?
Iron replacement tablets or iron-supplemented
vitamins should be taken only as directed by a physician to treat actual
iron deficiency or to
prevent iron deficiency in high-risk
circumstances (e.g. pregnancy). People with Thalassemia trait (Thalassemia
minor) are not having a grater risk of complications from iron in the diet
than anyone else in the general population. There are instances, however,
in which coincident conditions can increase the risk of iron overload.
For example, people with Thalassemia trait who also inherit the gene for
hereditary hemochromatosis can accumulate dangerous levels of iron by using
dietary iron supplements.
Can
the anemia produced by Thalassemia be corrected or improved by
taking more iron?
In the absence of concomitant iron deficiency, iron supplementation will neither correct nor improve anemia due to Thalassemia. For people with both iron deficiency and Thalassemia, iron replacement will lessen the severity of the anemia, until the iron deficiency is corrected. The blood count will level off and no further improvement will occur.
What
is the best treatment for a person with severe Thalassemia
(Thalassemia major or Thalassemia
intermedia) who accumulates too
much iron?
Excess iron accumulation is a leading cause of clinical deterioration and often death in patients with severe forms of Thalassemia. The excess iron can be removed from the patient’s body only by iron-binding drugs called chelators. The most widely used chelator is desferrioxamine, whose trade name is Desferal®. This medication can prevent many of the complications associated with iron overload. A special pump that the patient wears slowly gives the medication under the skin. Desferal® has been given as an intravenous infusion at the time of blood transfusion. This method of delivery does not effectively remove iron. Desferal® is not effective when given by mouth. A new oral agent called Deferipone or L1 continues to be tested in people. The drug effectively removes iron from many patients with substantial iron overload. The key unknown with this agent is its safety.
Consult your doctor when you see
signs and symptoms suggestive of anemia (Thalassemia) Inform your doctor
about your family history of
Thalassemia. Treatment should not
begin until your doctor will examine the results of laboratory tests. Make
sure your child maintains a well
balanced diet.
Over the past three decades, advances
in blood transfusions regimens and other medical research have allowed
patients with Thalassemia major to live relatively normal lives. Life expectancy
has been extended beyond the fourth decade of life often with minimal physical
symptoms. The number of affected individuals is increasing since carrier-screening
programs have not completely prevented new births and patients now live
longer. Most importantly, this growing population is dependent upon life-long
blood transfusions; in one year, 16,000 units of washed red blood cells
are used to treat Thalassemia patients in Canada. Thalassemia is a relatively
rare disorder in Canada so parents and patients have joined together to
form a notional network of Thalassemia groups. These provincial organizations
are entirely made up of volunteers who come from all walks of life and
many diverse cultures to work towards a cure for Thalassemia and a better
life for those already affected by this disease. The Ontario Thalassemia
Foundation is one such organization whose mission is to support and fund
medical research, treatment, patient services, public awareness and education.
Scientists and public health officials predict that with global improvements
in childhood disease prevention and treatment, and with targeted programs
to prevent mortality from malnutrition, Thalassemia will become a worldwide
issue in the next century. It is our hope that by providing electronic
education about the disease, we can raise awareness, encourage people to
get tested for trait, and spread knowledge about comprehensive treatment
to the global community.
![]()
Disclaimer: This research
and its content are provided here as information only. If you have any
questions about your health,please contact your health care provider. The
author presents this data as is, without any warranty of any kind, express
or implied and is not liable for mistakes, errors, or omissions.
News. For news check info regarding the new product Exjade made by Novartis, a Desferal substitute. Exjade will be taken with water. No pump and needle need.
Novartis link: http://www.novartis.com/
Thalasemia
Romanian Association
(Asociatia Persoanelor cu Thalasemie Majora) -is heaving the head office at : Romanian
Institute of Hematology and Blood Transfusion |
|
|
Romanian Thalassemia Association contact
information: Str. Constantin Caracas nr. 2-8, Sector 1 ,
|
Bank information:
|
|
January
30, 2006 http://www.webmd.com/content/article/118/112847.htm |
Every
year, about 8 million babies worldwide are born with gene-related birth defects,
says a new report from the March of Dimes.
That
figure amounts to 6% of all global births in a given year, the report shows.
There
are more than 7,000 genetic or partially genetic birth defects. Five common
types account for a quarter of the world's cases, says the March of Dimes:
·
Heart defects: More
than a million births worldwide yearly.
·
Neural tube defects
(including spina bifida): Nearly 324,000 births worldwide yearly.
·
Blood disorders (such
as sickle cell disease and thalassemia): More than 307,000 births worldwide
yearly.
·
Down syndrome: More
than 217,000 births worldwide yearly.
·
G6PD deficiency
(enzyme deficiency that causes anemia): More than 177,000 births worldwide
yearly.
More
than 3 million children die of genetic birth defects by age 5, and almost as
many may be permanently affected by their birth defect, the report shows.
|
January 8, 2006 http://www.tmcnet.com/usubmit/2006/jan/1269073.htm |
Thalassemia information:
www.Thalassemia.ca
Thalassemia Foundation of Canada
http://www.thalassaemia.org.cy/
Thalassaemia International Federation
http://www.Thalassemia.org/home/ie_op/h2.htm
Cooley's Anemia Foundation
http://www.abanet.it/fondazioneberloni/ing/
Berloni Foundation against Thalassemia
http://geocities.datacellar.net/HotSprings/5917/whatis.htm
Thalassaemia On-Line
http://www.healthlinkusa.com/Thalassemia.htm
Links to websites which may include treatment, cures, diagnosis, prevention,
support groups, email lists, messageboards, personal stories, risk factors,
statistics, research and more.
Amniocentesis and CVS:
www.melbuswomen.com.au
Melbourne Ultrasound for Women
http://www.dsav.asn.au/students/genetic_counselling.htm
Genetic Counselling & Pre Natal Testing
![]()
Please feel free to contact me:
|
Updated: |
|
![]()
![]()
![]()

