![]()
![]()
Alexander V. Spirov1,2 and Maria G. Samsonova1
1Institute of High-Performance Computing and Data Bases
2The Sechenov Institute of Evolutionary Physiology and Biochemistry
St Petersburg, Russia
The large-scale projects on human and several model organisms DNA sequencing lead to rapid growth of biological information. The well known Genbank and SwissProt databases contain essentially structural information. It is now necessary to design databases containing functional information that is focussed on specific problems of molecular biology. These will contain a broad spectrum of information including pictures, schemes and movies. The distinctive feature of these databases will be their ability to serve not only as an information depository but also as tools for derivation of new knowledge by means of computer analysis.
One of such specialized data bases is the GeNet database located on site: http://www.csa.ru/Inst/gorb_dep/inbios/genet/genet.htm, which is designed in the Bio-information Systems Lab of our Institute.
GeNet contains the information on functional organization of regulatory genetic networks acting at embryogenesis. The regulatory genes play a crucial role in embryo-genesis, controlling both activity of downstream regulatory genes (crossregulation), as well as their own activity (autoregulation). Most of genes of the network encode a transcription factors, which function is activation or repression of downstream target genes. In turn down-activated members of network switch on structural genes at the appropriate time and place. Thus the network of regulatory genes defines the genome activity during embryo development, and to reveal the mechanisms of embryogenesis it is necessary to understand the principles of regulatory genetic networks organization.
The hypertext version of GeNet is build on the basis of comparative evolutionary approach. The information in the database is presented in several categories: genes entries, regulatory regions entries, gene interactions entries, bibliography entries and graphical representation of gene interactions. Each gene entry contains as mandatory such fields as definition, expression pattern, sequence, regulatory regions, regulatory connections (upstream and downstream genes), evolutionary homologues, links to other databases, bibliography. The expression pattern field involves the images of expression pattern of segmentation genes in fruit fly Drosophila melanogaster. The work on incorporation in GeNet quantitative data on segmentation genes expression proceeds in collaboration with Dr. J.Reinitz (Brookdale Center of Molecular Biology, Mt.Sinai School of Medicine), whose group obtains gene expression data in experiments.
The regulatory element entry in GeNet contains such obligatory fields as definition, keywords, organism, bibliography, sequence and coordinates of sites for transcription factors binding. Gene interactions entries contain the target or effector gene name, mechanism of interaction, experimental proofs and bibliography. Graphical representation of gene interactions is accomplished in the form of flow diagrams, consisting of nodes and arrows, as well as in the form of the Java applets, (e.g., http://www.csa.ru/Inst/gorb_dep/inbios/genet/ Graph/genes.html) that permits to emphasize the interacting genes in the network, to reflect the mode of genes action, to drag genes for better visualization of links between them, etc.
Using information collected in GeNet we present the results of analysis of genetic networks controlling early development in fruit fly Drosophila.
The information in GeNet provides better understanding of molecular mechanisms of regulation of genes activity at embryogenesis. We use this information for to reveal the principles of organization of regulatory regions of genes controlling early morphogenesis stages in D. melanogaster, for to describe and analyze gap and pair-rule genetic networks, as well as for investigation of evolutionary conservation of these networks organization.
In brief, initial steps of embryo development in every organism can be described as follows (Lawrence and Morata, 1994; Jackle et al., 1992). In fertilized zygote there are maternally predetermined concentration gradients of several morphogens, which exponentially decrease with distance. The global challenge in early development is a conversion of this analogue input into digital one. This conversion is fulfilled by the system of macromolecular devices which is formed by genes controlling morphogenesis. The central role in this system play the coupled complexes of general transcription factors with promoters as well as specific transcription factors with cis-regulatory regions (enhancers and silencers). This of a sort molecular probes cooperatively (“all - none”) respond to exceeding threshold concentrations of transcription factors (Johnson and Krasnow, 1992). It is believed that such molecular probes can recognize at least two-fold change in transcription factor concentration (Schulz and Tautz, 1994). Essentially that the conversion of input is accomplished not by a single gene, but by the cascade of interacting genes.
The main function of genes controlling early morphogenesis stages (See legends to Figures 1 and 2) is "reading" and translation of maternal morphogen gradients into the parasegmental organization of early embryo (Driever and Nusslein-Volhard, 1988; Driever et al., 1989). Reading of maternal morphogen gradients is accomplished by high sensitivity of transcription initiation complexes to morphogens concentration thresholds. In turn this sensitivity is based on a multitude of maternal morphogen binding sites in the cis-regulatory regions of each gene, as well as on their differential affinity (Driever et al., 1989: Jiang and Levine, 1993; Simpson-Brose et al., 1994; Stanojevic et al., 1989). As the result each region along embryo's anterior-posterior and dorsoventral axis is characterized by a specific set of expressed genes.
Structure of Enhancers, Reading the Morphogen Gradients. There are the gap and the pair-rule genes which read the gradients of maternal morphogens BCD and HB along anterior-posterior axis. The binding sites of these transcription factors are found in quantity in many enhancers of gap and pair-rule genes and are substantially differentiated with respect to affinity. Table 1 shows the properties of 12 the most thoroughly characterised cis-regulatory elements of 4 gap, 2 pair-rule and 3 selector genes. It appeared that each cis-regulatory region contains on the average 15 - 20 binding sites for 5 transcription factors. The density of binding sites distribution is very high as one site falls within the region of 20 - 100 bp.
Table 2. Enhancers of Drosophila segmentation genes.
| Regulatory element | Length of element (in bp) | Transcription factors binding sites1 | |||||||||||||
| BCD_____ HB______ KR_____ KNI_____ GT_____ CAD____ TLL | |||||||||||||||
| hb anterior | 731 | 6 | + | 2 | + | 2 | - | ||||||||
| hb posterior | >1400 | p | - | 5 | ± | 8 | + | ||||||||
| Kr730 | 730 | 6 | + | 10 | + | 1 | - | 6 | - | 7 | - | ||||
| kni-enhancer | ~1000 | 4 | + | 10 | - | 6 | - | 1 | - | 12 | + | 8 | - | ||
| tll-enhancer | 350 | 8 | + | ||||||||||||
| eve stripe#2 | ~800 | 5 | + | 3 | + | 6 | - | 3 | - | ||||||
| eve stripe#3+7 | 508 | 11 | - | 5 | - | ||||||||||
| h stripe#5 | 302 | 4 | - | p | + | 1p | - | ||||||||
| h stripe#6 | 205 | p | - | 7 | - | 8 | + | ||||||||
| en 5'element | 100 | 2 | + | 1 | - | ||||||||||
| Ubx: PBX | 254 | 3 | - | ||||||||||||
| Ubx: BRE | 500 | 5 | - | p | - | ||||||||||
1p means “putative” binding sites, ‘+’ activation, ‘-’ repression.
The transcription factor HB controls the greatest number of regulatory elements - 11. Transcription factors BCD and KR regulate 7 elements each. Homeodomain protein BCD in 5 instances from 7 acts as activator, while Kr containing Zn-finger as a DNA-binding domain produces in all cases repression. HB protein which is highly homologous to KR acts just as activator so also repressor. Nuclear receptors KNI and TLL act in the same way. Homeoproteins (BCD, FTZ and CAD) appear more often to act as activators. The similarity of Kr and kni genes enhancers in the set of binding sites is evident.
In the whole regulatory elements presented in Table 1 contain 161 experimentally characterized binding sites for 9 transcription factors. More detailed analysis of regulatory elements sequences shows that 52 sites form 18 overlapping clusters. Among them 10 clusters consist of 2 overlapping sites, in 7 such cases one of sites binds activator, the other - repressor. Three clusters are formed by the overlap of 3 sites. The rest 5 clusters are composed from 4 or 5 partly overlapping sites.
Thus, summing up the data presented above, it is possible to conclude that regulatory elements of genes controlling early embryogenesis show striking similarity of their structure and function:
The networks of genes controlling embryo development in fruit fly D.melanogaster are among the most thoroughly studied (Jackle et al., 1992). Gentic cascades, each member of which controls expression of downstream target genes, are followed in Drosophila from maternal genes acting at oogenesis to genes controlling development of imago. These networks encompass up to hundred known at present genes, which code for transcription factors, transmembrane receptors and their ligands (Casares and Sanchez-Herrero 1995; Driever et al., 1989; Hoch et al., 1992; Margolis et al., 1995; Pignoni et al., 1992; Rivera-Pomar et al., 1995; Small et al., 1991; 1992; Pankratz et al., 1992). Despite of them, it is possible now to reconstruct the complete scheme of genetic network functional organization only for genes controlling early stages of development.
Gap Genetic Network. Gap genes act on the initial stage of the process of conversion of smooth exponential gradients of maternal morphogens along anterior-posterior and dorsoventral zygotic axes into discreet succession of embryonic para-segments, further pair-rule and selector genes take part in this process.
Gap genetic network (Figure 1) is formed by a small number of genes encoding transcription factors, which belong to Zn-finger, steroid-retinoid receptor and homeo-protein superfamilies. Gap genes activation and repression is triggered by exceeding the threshold concentrations of maternal morphogens, each gene being characterized by individual threshold values. These morphogens are transmembrane receptor TOR, which forms concentration gradients at the embryo termini, as well as BCD and HB, CAD and NOS, DL proteins, which gradients decrease correspondingly in the anterior-posterior, posterior-anterior and dorsoventral directions (Casanova and Struhl, 1993; Driever and Nusslein-Volhard, 1988; Pignoni et al., 1992; Simpson-Brose et al., 1994). It is noteworthy that practically all morphogens (BCD, HB, DL) may activate some genes and repress the other. It is also essential that several gap genes (Kr and hb) are autoregulated.
At the same time members of gap genetic network does not form closed activation circuits, which are characterized by interactivation of genes (eg. gene A activates gene B, B activates C, while C is the activator of A). It should be mentioned that such closed activation circuits are present in genetic networks controlling later stages of embryonic development.
Even cursory examination of gap genetic network scheme reveals that each gene is involved in many regulatory interactions and that negative regulatory links dominate (Table 1). At most thoroughly characterized genes hb, Kr, kni and tll regulate about from 7 to 10 other genes each, on average only two of them being activated. Such structure of genetic network leads to activation of no more than two gap genes in each particular region of sincytial blastoderm, which activity inhibits expression of other genes from this group.
We shall also underline now that terminal and trunk gap genes at blastoderm stage have usually two bands of expression, one of them located closer to the anterior, the other - to the posterior end of the embryo. Gene Kr is an exception, as its band of expression divides early embryo approximately on anterior and posterior halves. In some genes (eg. hb) each expression band is regulated by autonomous enhancer, analysis of other gene regulatory regions up till now does not reveal such one-to-one correspondence (e.g., it seems that one regulatory element controls both bands of kni gene expression).
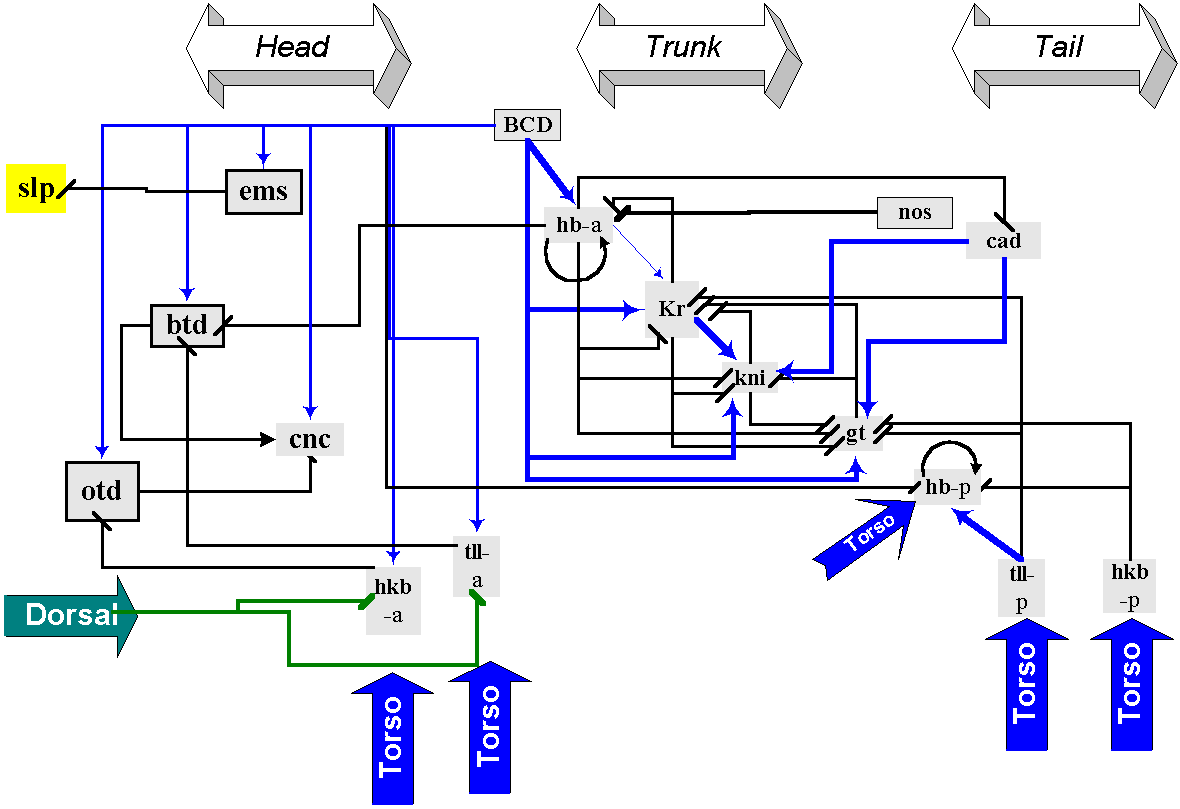
Figure 1. Genetic network defining gap gene expression domains in head, trunk and tail regions of the blastoderm embryo. Maternal morphogen BCD activates both head gap genes ems, btd, cnc and otd, and trunk gap genes hb (anterior element), Kr, kni and gt. Head gap gene btd activates, in turn, another member of this group cnc, while otd represses it. Maternal and zygotic CAD activates kni and gt. hb activates and represses Kr (in concentration-dependent manner) and represses the anterior limits of both kni and gt expression, while Kr activates kni and represses gt. Torso cascade activates terminal gap genes tll and hkb, as well as anterior element of the hb gene and head genes otd and cnc. Maternal morphogen DL represses tll and hkb in the anterior pole. Arrows indicate positive regulation; lines ending with a vertical bar designate negative inputs. “-a” marks regulatory elements, controlling anterior bands of gene expression; “-p” marks regulatory elements for posterior bands of expression.
Pair-rule Genetic Network. The next step in conversion of maternal morphogen gradients into segmental organization of early embryo is accomplished by pair-rule genetic network (Reinitz and Sharp, 1995; Small et al., 1991; 1992). The primary pair-rule genes are activated firstly as the result of maternal morphogens and gap genes products action. The activity of primary pair-rule genes is necessary for activation and maintenance of secondary and tertiary pair-rule genes. Pair-rule genes express in a series of seven stripes along anterior-posterior axes of early embryo.
Several distinctive characteristics of gap genetic network are more distinctive in the case of pair-rule genetic network. As in the case of the gap genetic network the most thoroughly studied primary pair-rule genes even-skipped (eve) and hairy (h) are involved in many regulatory interactions with other genes. The activity of these genes is regulated by maternal morphogens, gap genes as well as by pair-rule genes themselves (Figure 2). Altogether the total number of regulatory interactions may be as much as 12, with only 2 or 3 being activations.
As in the case of several gap genes each stripe of primary pair-rule genes expression is controlled by autonomous enhancer (Table 1). Each enhancer contains binding sites both for activators and repressors. A great number of regulatory interactions in which pair-rule genes are involved may be ascribed to the necessity of control of each of these genes expression in seven different embryos regions, which differ in concentrations of maternal morphogens and gap gene products.
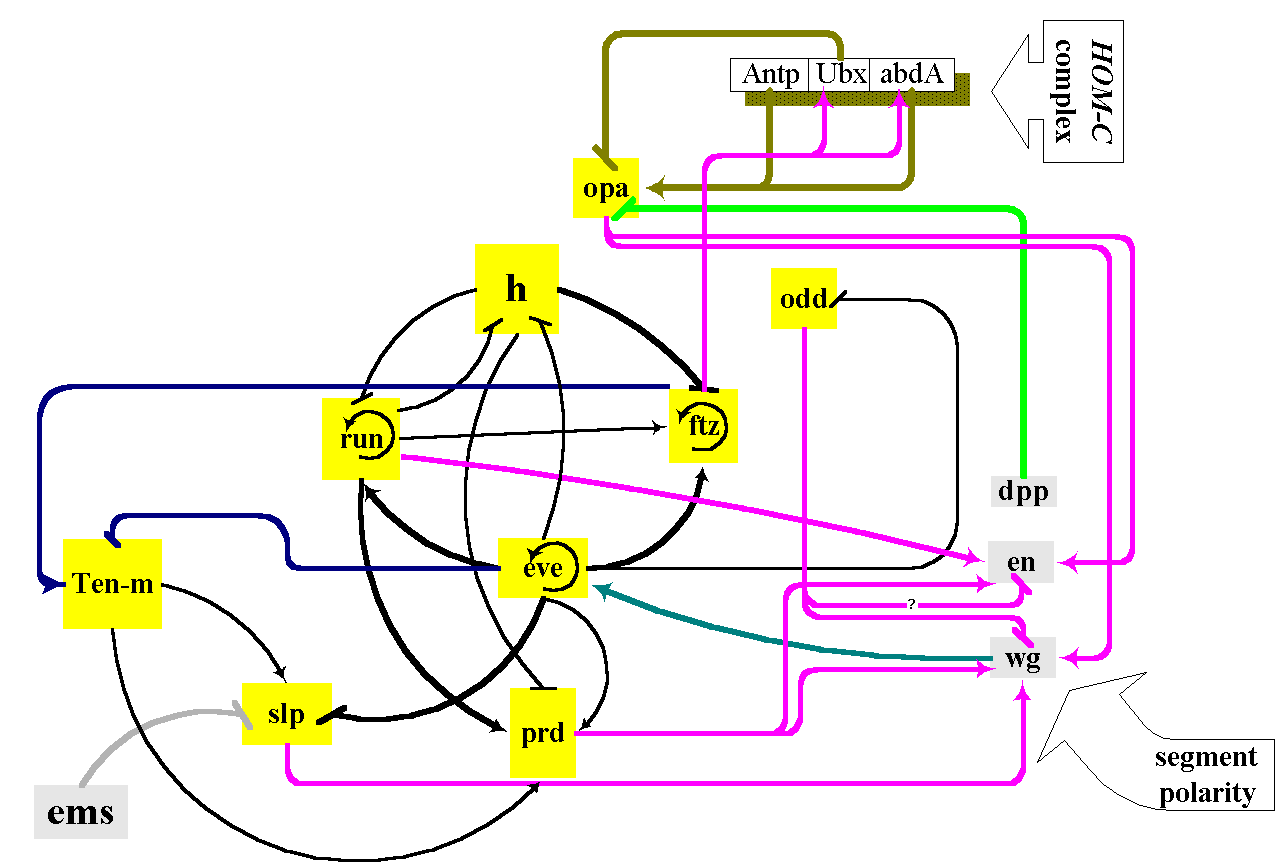
Figure 2. Functional relations of members of genetic network controlling pair-rule genes expression patterns in the blastoderm of early embryo. Initially the expression of primary pair-rule genes is controlled by maternal morphogen BCD an maternally and zygoticaly expressed HB and CAD. Pair-rule gene cross-regulation exhibits hierarchical structure prior to gastrulation. One pair-rule gene eve is not regulated by other pair-rule genes. Two others, h and run, are regulated by eve, but not by other pair-rule genes. These three are known as primary pair-rule genes. Another pair-rule gene, ftz, is regulated by eve, h and run but not by other pair-rule genes. ftz and odd are considered as secondary pair-rule genes. prd and slp are regulated by all other pair-rule genes and therefore are considered to be tertiary pair-rule genes. In turn pair-rule genes control expression of segment polarity (wg, en) and homeotic (Antp, Ubx, AbdA) genes.
Several secondary pair-rule genes (eg. ftz) show lack of multiple enhancers each controlling separate stripe of expression pattern and seems to be under control of more limited genes number.
It should be pointed out that autoregulation is typical of pair-rule genes. eve gene autoregulatory element is one of the most thoroughly studied among autoregulatory elements in Drosophila (Small et al., 1991).
The study of Boolean network models by S. Kauffman with co-workers over last thirty years leads to formulation of fundamental features of genetic networks global behaviour (Kauffman, 1993). On the other hand at present a wealth of experimental data permits us to understand in details the functioning of genetic networks controlling early development in Drosophila. Thus we can interprete these data in form of Boolean network models for comparison of behaviour of theoretical networks and the real biological ones.
A Boolean network is a system of interconnected binary elements and any element in the network can be connected to a series of other elements. Each individual element uses a logical (Boolean) rule to compute its value based on the values of the other elements it is connected to. The state of the system is defined by the pattern of ON/OFF states of all of its elements (For Introduction See Somogyi and Sniegoski, 1996).
One of the key features of the Boolean networks is that all states, i.e., ON/OFF pattern of its elements at a particular time point, lead to or are the part of an attractor. The attractor is a distributed structure, based on the state (a point attractor) or series of states (a dynamic attractor) which repeats on itself. All the states leading to or being a part of this attractor, constitute the basin of attraction.
Each network must reach one of several possible attractors depending on the initial conditions. Any state within a particular basin of attraction can be switched to any other state within this basin, without changing the global characteristics of the system. Despite the general resistance of attractors to point alterations in states, the boundaries of each basin of attraction must have at least one state, in which a single point alteration will determine which basin the system will fall into.
Let us illustrate the prospects of modelling of Drosophila segmentation networks in framework of Boolean model. Considering the data of GeNet database we formulate the Boolean rules for segmentation genetic networks as follows: action of at least one repressor switches off a target gene, while activation by at least one activator switches the target gene on, provided that a repressor is absent.
We perform the analysis of segmentation genetic networks by means of Discrete Dynamics Lab package developed by Dr. Wunsche.
Modelling of Head Segmentation Genetic network. The head segmentation genetic network consists of 3 genes encoding maternal morphogenes, Hb, which is both maternal and zygotic gene and is presented in network by its anterior enchancer hba, 5 genes encoding head gap genes, 2 anterior enchancers tlla and hkba of terminal genes and trunk gene Kr (Figure 1).
It should be pointed out that correct modelling of expression pattern of head segmentation network in framework of Boolean model requires elimination of negative link from gene Kr to slp, as its presence leads to inactivation slp in all attractors. However, the inclusion Kr in head segmentation genetic network is mandatory, as elimination of this gene profoundly alters a whole picture of network dynamics as compared with experimental data.
The basin of attraction for all possible states of head segmentation genetic network consists of 9 point attractors (Table 2).
It turns out that some of these attractors may be correlated with definite region of embryo head on blastoderm stage. The 1st attractor "all off" reflects the trivial fact that in the absence of maternal morphogens inputs all elements of genetic network will be switched off. The other attractors are characterized by combined activity of head gap genes and morphogens.
Attractor III is formed by cnc and terminal gap genes, while attractor IX is characterized by activation Hb in addition to these genes. Earliest expression cnc in the late blastoderm is found in labial and mandibular parasegments (Grossniklaus et al., 1994; Mohler et al., 1995; Pignoni et al., 1992). Hence attractors III and IX may correspond to these parasegments.
ems alone and in combination with DL locally represses slp in two adjacent regions. The so-called ventral repression depends on DL in conjunction with ems, while splitting of initially single band slp expression in two bands is under control ems only (Grossniklaus et al., 1994). As we can see, the attractor VII, characterized by activity btd and ems, but not slp could correspond to one of these two regions. On the other hand, attractor VI and VIII, formed by otd, ems, cnc, Kr, BCD and DL genes may correspond to ventral part of prospective ocular parasegment, where slp activity is repressed by ems and high DL concentration (Cf. Mohler et al., 1995; Walldorf and Gehring, 1992).
Attractor V, characterized by activation slp, ems, btd, hkb and Kr genes in presence BCD may correspond to the region of combined expression slp, ems and btd genes, which defines the morphogenesis of intercalary segment (Grossniklaus et al., 1994).
At last, the most anterior domains in a head region are marked by otd gene expression (Finkelstein and Perrimon, 1990). Attractor IV may mark this zone. Data of mutation analysis suggest that domain solely expressing otd does not correspond to any head segment.
Table 2. Basin of attraction field of Drosophila head-gap genetic network.
| No | Switched-on genetic elements | per-cents from all possible states | Number of layers |
| I | - | 12.5 | 4 |
| II | DL | 12.5 | 4 |
| III | cnc, tlla, hkba, TOR | 10.9 | 2 |
| IV | cnc, otd, DL, TOR | 12.5 | 4 |
| V | slp, ems, btd, hkba, Kr, BCD | 12.5 | 5 |
| VI | cnc, otd, ems, Kr, BCD, DL | 12.5 | 3 |
| VII | btd, ems, hkba, Kr, BCD, TOR | 12.5 | 5 |
| VIII | cnc, otd, ems, Kr, BCD, DL, TOR | 12.5 | 3 |
| IX | cnc, hba, tlla, hkba, TOR | 1.6 | 2 |
Thus, in spite of imperfect knowledge on mechanisms of cis-, trans- regulation of head gap genes activity, we get a reasonable correspondence of attractors to the regions of cell fate determination in the limits of at least four head segments.
S. Kauffman investigated the characteristics of Boolean nets that correlate with the appearance of spontaneous order, namely, the connectivity and the homogeneity. Order arises for networks with connectivity k=2 or in networks characterized by higher connectivity and the parameter P value, which measures the internal homogeneity, greater then some critical value Pc.
The evaluation of head segmentation genetic network parameters shows that
the average value of P is equal to 0.65,
each element of network has in average 2.5 functionally linked neighbours,
totally, the number of positive links is nearly equal to the number of negative links (17 and 13 correspondingly),
one more feature of the net is the low branching level for all attractors (maximum 5).
Moreover, clearly defined hierarchical structure is inherent to head segmentation genetic network: the initial input of 4 morphogens activates 9 target elements. These downstream elements are interconnected by a number of negative links.
Analysis of head segmentation genetic network stability towards removal of elements confirms the hierarchy of network organization. The morphogens removal (i.e. BCD) decrease the attractor number to 5, while 3 of them are new. On the contrary the removal of downstream targets of morphogens action (the 2nd level of network organization) does not substantially change attractors number, but modifies their structure. For example Kr gene exclusion leads to elimination of the 4 and appearance of the 7 new attractors, while 5 other attractors do not change. Thus the resistance of head segmentation genetic network to elimination of elements depends on weight of their input into network organization.
In comparison with the head segmentation genetic network the random Boolean network, characterized by the same neighbourhood k=2.5 but P value equal to 0.5, reaches quite different attractors. Hence, this comparison shows the peculiar features of head segmentation network. The basis for these features is in the fact that the head segmentation genetic network was formed in the process of evolution over ten millions of years (Patel, 1994).
It becomes evident now that a cell function cannot be understood in terms of ''one gene - one function'' paradigm. For to solve this problem it is necessary to understand how genetic networks operate.
To this end it is necessary to perform large-scale measuring of gene expressions spectra, to design databases containing gene expression data and to develop program tools to infer the information on genetic networks functioning on the basis of expression databases data.
The GeNet database is intended as a data base of new type oriented on representation of results of analysis of genetic networks structure, function and evolution. Now it contains the images of expression pattern of segmentation genes in fruit fly D.melanogaster. The inclusion of these images is the first step towards design of quantitative atlas of Drosophila segmentation genes expression, containing both 3D and numerical data.
The information collected in GeNet can be used for derivation of new knowledge on genetic networks functioning by means of computer analysis.
The head segmentation network is relatively autonomous part of Drosophila embryo segmentation network. At present most of functionally essential regulatory links in these networks are revealed. Most of them consist in interaction of trans-acting proteins with cis-regulatory regions.
Good understanding of mechanisms of gene interactions permits to model the information processing in head segmentation network by means of Boolean network theory.
The unique feature of segmentation genetic networks is the presence of several maternal morphogens, which are outputs of other genetic networks. Thus in basin of attraction for all possible states all combination of morphogens can be switched on. However only attractors with natural combination of morphogens reflect the actual pattern of genes activation in networks.
Boolean models of segmentation networks substantially differ from random Boolean network. These networks have hierarchical structure. They are sensitive to elimination of elements. Moreover the degree of disturbance elicited by elements elimination depends on the weight of element in network organization.
Supported by Russian Foundation for Basic Researches (Grant No 96-04-49350).
![]()
![]()
This page hosted by ![]() Get your own Free Homepage
Get your own Free Homepage