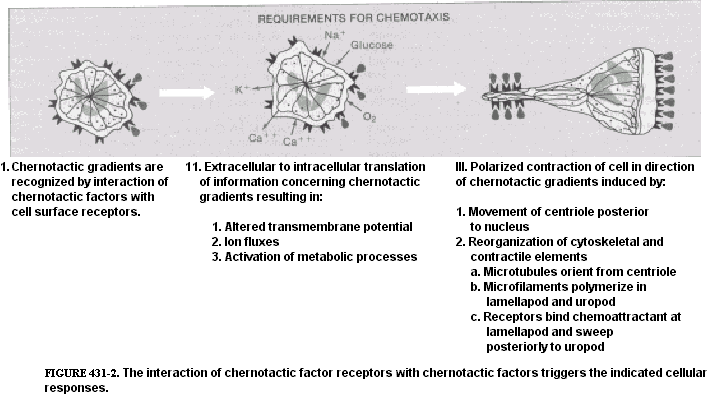
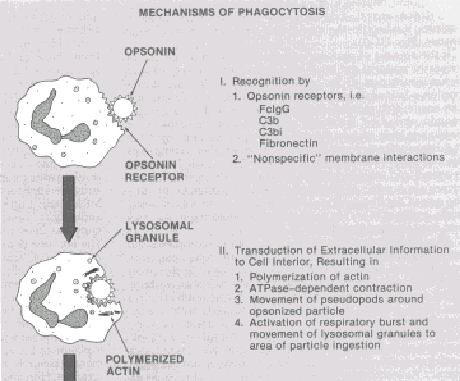
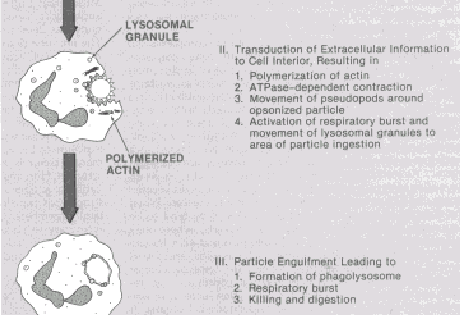
|
Immunologic processes mediate the localization and destruction of substances that, if disseminated, could disrupt the host's complex internal milieu. The immune system has several unique features that permit it to combat microbial invasion and provide resistance against the spread of cancer. Unlike *other tissues, the immune system consists not only of fixed structures (i.e., thymus, spleen, and lymph nodes) but also of motile cells that wander throughout the body, performing surveillance. It is also the only tissue able to destroy other components of the host. Both the protective and destructive abilities of immunologic processes relate largely to their inflammatory potential. Understanding how inflammation is initiated is thus essential for understanding the mechanisms of immunologically mediated resistance and for comprehending how tissue destruction occurs in the rheumatic disorders. To fulfill its function of host defense, the immune system must differentiate self from nonself and then rapidly destroy substances recognized as nonself. A progression of immunologic recognition, amplification of the immune reaction, accumulation of inflammatory cells, and finally destruction of the inciting agent is an ongoing subclinical process. Inflammatory reactions can be initiated by either specific or nonspecific medns (Table 431-1). Recognition of unique determinants (epitopes) on antigens by antibodies or by receptors on lymp~ocytes initiates immunologically mediated inflammation. Nonspecific recognition can be initiated by components of the immune system that bind to materials based on their charge, hydrophobicity, or lectin composition. Nonspecific recognition is mediated in part by phagocytic cells, such as polymorphonuclear leukocytes and macrophages, as well as by C3b, an initiator ofthe alternative pathway of the complement system (see Ch. 418), and by Hageman factor.
Following recognition of nonself, amplification systems are activated and lead to the production of mediators of inflammation. The type of amplifier involved depends upon the recognition component and the nature and location of the inciting material. Amplification components of the immune system such as complement cleavage products, cytokines, and other phlogistic factors magnify the initial response to nonself Infla atorv reactions can also be initiated bv non-immunologic means. For example, inflammation following tissue necrosis results from the direct cleavage of complement components by lysosomal proteases released by injured cells. This phenomenon may play a role in extending cardiac: tissue damage following myocardial infarction. In gout or pseudogout, inflammation follows the phagocytosis, by polymorphonuclear leukocytes, of monosodium urate or calcium pyrophosphate dihydrate crystals. Ingestion of these agents by leukocytes leads to the release of lysosomal hydrolases as well as the production of chemotactic factors, which attract other inflammatory cells into the joint.
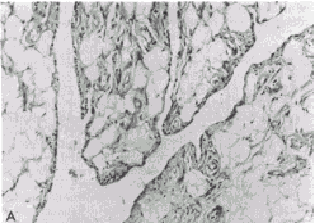
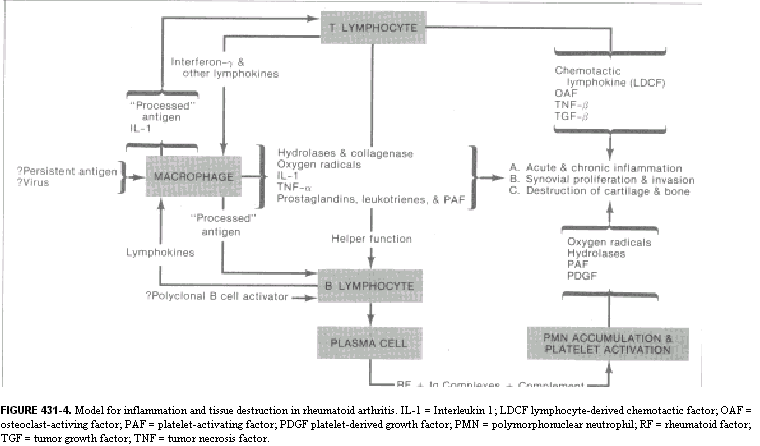
Regardless of the type of inflammatory response, the accumulation of granulocytes and macrophages can result in phagocytosis and degradation of the material that initiated the inflammatory event. The factors that determine whether an inflammatory response will be protective or destructive depend in part upon the nature and location of the inciting agent, its quantity, digestibility, the genetic makeup, and the immunoregulatory competency of the host. In general, when the antigen or other inciting agent is rapidly disposed of, the inflammatory process is self-limited. When antigen persists or is excessive in amount, the inflammatory response can be locally destructive and become clinically apparent. RHEUMATOID SYNOVITIS AS A MODEL OF INFLAMMATORY-MEDIATED TISSUE DESTRUCTION A chronic, locally destructive inflammatory reaction in humans is exemplified by the synovitis present in some connective tissue disorders. The prototype disease is rheumatoid arthritis. The diarthrodial joint has several features that influence inflammatory processes that occur there. The synovial membrane is highly vascular and lines all intra-articular structures, except for cartilage. The synovial lining is devoid of a basement membrane and thus permits relatively free diffusion of soluble substances. Moreover, the synovium lines a closed cavity, the joint space; therefore any reactive materials gaining entrance to the joint space are difficult to remove. Normal synovium is a thin layer of tissue whose lining is composed of two principal cell types supported by a loose connective tissue stroma (Fig. 431-1A). The Type A cell is rich in surface pseudopodia, and its cytoplasm has many lysosomes and prominent Golgi complexes but little rough endoplasmic reticulum. Type A synoviocytes have many characteristics of macrophages and are phagocytic, and ultrastructural studies have implied a secretory role for these cells as well. The Type B synoviocyte exhibits prominent rough endoplasmic reticulum but few vacuoles, lysosomes, or cell processes. This cell is primarily a secretory cell, hyaluronic acid being an important product. Synoviocytes may, however, be multipotential, with their morphology reflecting the net result of stimuli present in the local milieu. Beneath the synovial membrane there are collagen fibrils, fatty tissue, and an extensive capillary network. Fibroblasts in this area produce Types I and III collagen. Lastly, the dense fibrous joint capsule provides support for the synovial lining membrane and separates the articular space from surrounding structures. Rheumatoid synovitis exhibits three main components: inflammation, proliferation, and infiltration. In early stages, both Types A and B cells proliferate and increase in size. Fibrin deposits are frequently present on the inner synovial lining. Polymorphonuclear leukocytes predominate in the synovial fluid exudate, but cells are seen as infiltrates only in the superficial synovial layer. The supporting stroma beneath the lining cell layer becomes edematous and develops an increase in the number of small blood vessels. Concurrently, there are focal accumulations of inflammatory cells consisting of lymphocytes, plasma cells, macrophages, and occasionally mast cells. If the inflammatory synovitis persists, a proliferative lesion develops, characterized by synovial membrane thickening and projection of villous formations into the articular cavity (Fig. 431-1B). there is a concomitant increase in supporting connective tissue, small blood vessels, mononuclear cell infiltrates, and occasional multinucleated giant cells, as well as increased numbers of undifferentiated mesenchyme-like cells that have both phagocytic and synthetic potential. Clusters of poorly differentiated synovial cells may be found invading cartilage and subchondral bone. As the proliferative lesions progress, fibrous-mesenchymal tissue (pannus) begins to invade and replace cartilage and bone at the periphery of the synovial reflection (Fig. 431-1C). The invasion of cartilage, subchondral bone, and tendon by inflammatory synovial tissue results in collagen destruction, degradation of proteoglycans in the cartilage matrix, and bony resorption. MEDIATORS OF INFLAMMATION THAT PARTICIPATE IN THE RHEUMATIC DISEASES Inflammatory reactions result from the local production of a number of mediators derived from humoral or cellular sources. A complex interplay of activation and suppression mechanisms modulates the type and magnitude of the inflammatory response. LIPID MEDIATORS. Phospholipids are major constituents of cell membranes, including those of leukocytes and platelets, and are subject to degradation by cellular phospholipases under certain conditions such as exposure of cells to inflammatory or noxious stimuli. Cleavage of phospholipids results in the release of arachidonic acid, which can be further metabolized into a number of biologically potent mediators and modulators of inflammation. Prostaglandins (PG) are synthesized from arachidonic acid following the action of the enzyme cyclo-oxygenase, which forms PGG, which is then reduced to PGH,. Depending upon the isomerase enzymes present in the particular tissue, prostaglandins (PGE,, PGF,,), thromboxanes, or prostacyclins will be formed. Leukocytes and explants of rheumatoid synovia produce predominantly PGE,, platelets produce thromboxane A, and endothelial cells produce prostacyclin. Thromboxanes are vasoconstrictors, whereas prostacyclins are vasodilators. PGE, appears to modulate a number of inflammatory events. It enhances vascular permeability, is pyrogenic, and increases sensitivity to pain. PGE, also stimulates formation of cAMP in many types of inflammatory cells and thereby suppresses a number of immunologic responses, including release of mediators from mast cells, lymphocyte blastogenesis, and lymphocyte-mediated cytotoxic reactions. An important source of PGE, in immunologic reactions is the macrophage. Supernatant fluids from explants of rheumatoid synovium stimulate bone resorption by enhancing osteoclast activity and release of bone calcium. This phenomenon is probably mediated in large part by PG, since it is inhibitable by indomethacin, which blocks their formation. Arachidonic acid can also be metabolized into another class of biologically active derivatives by the enzyme lipoxygenase. The hydroxy-eicosatetraenoic acids (HETEs) and the derivatives of 5-hydroperoxy-eicosatetraenoic acid (termed leukotrienes) are examples of these arachicionic acid metabolites and are synthesized by granulocytes, macrophages, and basophils. 5,12-HETE, also termed leukotriene B, (LTB,), is a potent chernotactic factor, whereas leukotrienes C and D stimulate bronchoconstriction. LTB, has been identified in the synovial effusions of patients with rheumatoid arthritis and ankylosing spondylitis. Large amounts of LTB, are produced when granulocytes phagocytize monosodium urate crystals, and production of LTB, is inhibited by colchicine. This chemoattractant may, therefore, be important in the pathogenesis of gouty inflammation. Platelet-activating factors (PAF) are a group of acetyl-alkylglycerol ether analogues of phosphatidylcholine. PAF causes platelet aggregation and is a potent leukocyte activator and chemoattractant. Leukocytes produce PAF following their stimulation by inflammatory mediators. BIOLOGICALLY ACTIVE AMINES. Histamine and Serotonin. Histamine is derived from the decarboxylation of histidine by the enzyme L-histidine decarboxylase. The majority of histamine is stored in mast cells and basophils and is complexed with mucopolysaccharides such as heparin. Stimulation of mast cells and basophils by a number of mechanisms causes secretion of histamine. This agent has diverse biologic activities, including constricting smooth muscle, enhancing vascular permeability, depressing leukocyte chemotaxis, blocking T lymphocyte function, and depressing further histamine release from mast cells and basophils. Histamine thus may modulate both acute and chronic inflammatory responses. Serotonin (5-hydroxytryptamine) is produced by the decarboxylation of 5-hydroxytryptophan. More than 90 per cent of body stores of serotonin are found in the gastrointestinal tract and the central nervous system; the remainder is present in the dense granules of platelets, The biologic role of serotonin in inflammation is not well understood, but it enhances the chernotactic responses of leukocytes and increases fibroblast growth in vitro. It also stimulates collagen formation. BIOLOGICALLY ACTIVE PEPTIDES. Complement Cleavage Products. The complement (C) system functions as an important amplifier of inflammatory events initiated by immunoglobulins IgG and IgM as well as inflammatory reactions initiated by release of hydrolytic enzymes from traumatized cells or by leukocytes. The biology and biochemistry of this complex series of proteins are described in Ch. 418. Two complement cleavage products, C3a and C5a, derived from the third and fifth C components, respectively, are mediators of inflammation in rheumatic disorders such as rheumatoid arthritis and will thus be described in greater detail here. C3a: Enzymatic cleavage of the a. chain of C3 by the earlieracting C components or by other proteases releases C3a, a peptide consisting of 77 amino acids. C3a mediates a number of biologic responses, including smooth muscle contraction, vasodilatation, enhanced vascular permeability, the degranulation of mast cells and basophils, and the secretion of lysosomal enzymes by leukocytes. C3a also has immunoregulatory effects and suppresses humoral immune responses in vitro by affecting T lymphocytes. C3a is the most abundant of the C peptides released upon activation of C in serum. The COOH-terminal arginine of C3a is required for its biologic activity, and cleavage of this amino acid by a carboxypeptidase-B-like enzyme in serum renders the peptide inactive. C5a: C5a has a number of structural and biologic similarities to C3a. C5a consists of 74 amino acids, the COOH-terminal constituent also being arginine. C5a is derived from cleavage of the ot chain of C5. In addition to having all the biologic activities of C3a, C5a is also an extremely potent chemoattractant for polymorphonuclear leukocytes, monocytes, and macrophages. C5a is the major source of chernotactic activity generated in serum treated with immune complexes or endotoxin and is also an important source of chernotactic activity in rheumatoid synovial fluids. In contrast to C3a, C5a potentiates humoral immune responses in vitro. Cleavage of the terminal arginine of C5a by a serum carboxypeptidase-B markedly diminishes its biologic activity. Crystal-Induced Chemotactic Factors. Leukocytes accumulate in the-synovial fluid of individuals with gout or pseudogout following the ingestion by neutrophils of monosodium urate or calcium pyrophosphate dihydrate crystals, respectively. Incubation of neutrophils with these crystals in vitro results in the production by the cells of LTB, and a polypeptide chemoattractant (CCF) with a molecular weight of 8400. The production of these chemoattractants is blocked by colchicine. A mechanism by which colchicine abrogates acute gouty arthritis may be its ability to inhibit the synthesis of chemoattractants by neutrophils. Kinin-Forming System. An intimate association exists between the activation and regulation of the intrinsic clotting, fibrinolytic, and kinin-forming systems. Hageman factor (HF) (Factor XII of the clotting system) is central to the activation of all three systems. HF is activated nonspecifically by a number of agents, including exposure to crude preparations of collagen, vascular basement membranes, monosodium urate crystals, calcium pyrophosphate crystals, and endotoxin. Negatively charged surfaces also activate HF. Upon activation, HF (an 80,000-dalton P globulin) is cleaved, and its active form HF, initiates the conversion of Factor XI of the clotting pathway to XIa, and the conversion of prekallikrein to kallikrein. Kallikrein activates plasminogen, an enzyme important in fibrinolysis. Kallikrein also cleaves serum kininogen to form bradykinin, a nonapeptide with potent biologic activities. Cl esterase inhibitor (ClINH), a protein that inhibits activated Cl, is also an important inhibitor of HF, and kallikrein. Bradykinin and two other kinins produced by tissue kallikreins from kininogen induce smooth muscle contraction, increase vascular permeability, and induce pain. Cleavage of fibrinogen by plasmin results in production of a number of products, including fibrinopeptide B, which potentiates the action of bradykinin and has chemotactic activity. Interleukins and Other Cytokines. Interleukins are immunoregulatory molecules synthesized by mononuclear leukocytes. Stimulation of macrophages by antigens as well as by factors from lymphocytes initiates the secretion of interleukin 1 (IL-1), a 15,000-dalton peptide with diverse biologic activities. Macrophages are required for many of the activities of lymphocytes, and certain of the "helper" functions of macrophages are mediated by IL-1. IL-1 may be identical to a factor termed mononuclear cell factor (MCF), which stimulates synovial cells to produce collagenase. IL-1 may also be identical to leukocytic pyrogen. Tumor necrosis factor-a (TNF-(x) is a macrophage product with IL-l-like activities plus the ability to kill many types of tumor cells (see Ch. 257). Interleukin 2 (IL-2), previously termed T-cell growth factor, is a 15,000-dalton polypeptide produced by T lymphocytes, which stimulates the continuous proliferation of activated T lymphocytes in culture. Transforming-growth factor-P (TGFP) is a 25,000-dalton homodimeric protein found in platelets and produced by stimulated lymphocytes. TGF-P inhibits further lymphocyte division and regulates differentiation of many cell types including fibroblasts. lnterferon--~ (IFN--~) is a 50,000 dalton protein produced by stimulated lymphocytes. IFN-,y activates macrophages and synergizes with other immunomodulators. LYSOSOMAL ENZYMES. Lysosomal enzymes are contained in subcellular organelles termed lysosomal granules. These enzymes degrade complex macromolecules. Lysosomal granules also contain antimicrobial constituents such as myeloperoxidase and lactoferrin. Leukocytes contain several types of lysosomal granules, two of which (primary and secondary) are differentiated by their staining characteristics. Lysosomal enzymes digest antigens following phagocytosis. However, since these enzymes may also be released during phagocytosis or upon cell death, they can cause tissue destruction. Lysosomal proteases found in leukocytes include collagenase, elastase, cathepsin D, cathepsin G, and gelatinase. These enzymes are capable of destroying extracellular structures and may participate in mediating tissue injury in the rheumatic diseases. Cathepsin D cleaves cartilage proteoglycan, whereas granulocyte collagenase is active in cleaving Type I and, to a lesser degree, Type III collagen of bone, cartilage, and tendon. Substrates of granulocyte elastase include collagen crosslinkages and proteoglycans, as well as elastin components of blood vessels, ligaments, and cartilage. Lysosomal hydrolyases also produce mediators of inflammation through their direct action on C components such as C5. Leukocytic hydrolyases can liberate kinin from kininogen. Plasminogen activator, an enzyme that converts plasminogen to plasmin (which stimulates fibrinolysis) is found in both granulocyte and macrophage lysosomes. Rheumatoid synovial collagenase is usually present as an inactive lysosomal proenzyme that requires plasminogen activator for conversion to its active form. Regulation of tissue destructive potential of the lysosomal proteases is mediated by protease inhibitors such as a, macroglobulin and a, antiprotease. These antiproteases are present in serum and in synovial fluids and inhibit proteases by binding to them and covering their active sites. PHYSIOLOGIC MECHANISMS OF INFLAMMATORY CELL ACCUMULATION The accumulation of inflammatory cells at sites of antigen is central to the inflammatory process. Polymorphonuclear leukocytes and macrophages are motile cells that have many common physiologic characteristics. Both can perceive gradients of chemoattractant molecules and migrate directionally along such gradients. They also perform endocytosis (ingestion), secrete lysosomal enzymes, and generate superoxide anions (see Ch. 148). Polymorphonuclear leukocytes and macrophages have specific surface receptors that perceive C5a, CCF, and LTB, as well as synthetic polypeptide chemo tactic factors . Binding of chemoattractants to the surface of phagocytes results in orientation of the cells toward the source of the chemotactic gradient. The cells lose their round configuration and become triangular, with the base of the triangle facing toward the chemoattractant gradient (Fig. 431-2). This change in cell shape requires rearrangement of intracellular cytoskeletal elements. Microtubules provide a front-to-back polarization, whereas actin filaments accumulate at the front and back of the cells and provide the contractile forces required for movement. Attachment of leukocytes to the vascular endothelium and other surfaces is a requirement for cellular motility. Chemoattractants stimulate the appearance of several adhesive proteins on the surface of phagocytes. These proteins include the iC3b receptor (mac 1, mo 1), LFA 1, and glycoprotein 150/90. These proteins are heterodimers that serve a number of adhesive functions. They consist of individual ot-chains and a common P-subunit. Their appearance on the surface of phagocytes enhances the cells' binding to endothelial cells, tumor cells, and other surfaces. Deficiency of these proteins is associated with a severe defect of leukocyte function and a marked increase in susceptibility to infection. Chemotactic factors can also initiate other cellular responses by leukocytes, such as superoxide anion production and lysosomal enzyme secretion. The concentration of chernotactic factors required to initiate these latter processes is approximately 10-fold greater than that required for the induction of chemotaxis. Thus, release of potentially toxic products from the cells may not occur until they arrive at the inflammatory site where the concentration of chemoattractants is high. |