|
TABLE 437-3. RAYNAUD'S
PHENOMENON IN MUSCULOSKELETAL DISEASE*

Selected immune events, other
than the autoimmune antinuclear antibodies, point also to the vascular
structures. Three of the four defined antigens to which SSc patients
show enhanced immune responsiveness are type I collagen, type IV collagen,
and laminin (the fourth being topoisomerase I), all three components
of blood vessels and the second and third being specific for basal lamina,
of which the endothelial basement membrane is a prototype. Could a heightened
immune response to basement membrane antigens select those persons destined
to develop SSc and perpetuate the disease in these immunogenetically
selected persons? As in the other rheumatic or diffuse connective tissue
disorders that show manifestations of autoimmunity, antinuclear antibodies
are prominent in SSc. With the recent introduction of rapidly proliferating,
human cell substrates (particularly the human laryngeal carcinoma cell
line, HEp-2), greater than 90 per cent of SSc patients have circulating
antinuclear antibodies in significant titer. The most sensitive and
specific of these is the anticentromere antibody (ACA) pattern, a B
cell immune response to a major organizing structure of the chromosome
called the centromere. ACA patterns are clinically useful in detecting
the limited cutaneous type of SSc. Antibodies to a soluble nuclear isomerase,
topoisomerase I, are currently emerging as the newest autoimmune serologic
"find" in SSc; both clinical and biologic relevance remains to be determined.
THE PATIENT.
Diffuse SSc. The usual age of onset is the fourth decade but may
range from the first to the eighth. There is no sex, race, or geographic
predilection. The onset may be abrupt and may present as swollen hands,
face, and feet associated with Raynaud's phenomenon (episodic pallor
of the digits, nose, or ears following cold exposure or stress associated
with cyanosis and followed by erythema, suffusion, tingling, and pain).
Fatigue is common; overt weakness may be present. The skin reveals a
nonpitting fullness, an inability to pinch skin folds, and the loss
of skin lines and creases in involved areas. These changes may evolve
over 12 to 18 months to include the fingers, hands, forearms, arms,
face, thorax, and abdomen, as well as the toes, feet, legs, and thighs.
The fingers and toes may be dusky or overtly cyanotic and are usually
cool to the touch. Blood pressure and pulse may be elevated, and evaluation
of swallowing, breathing, urinary excretory, and cardiac functions may
reveal abnormalities. Patients with diffuse SSc should be followed closely
for visceral involvement (see Table 437-2 and Fig. 437-1). Limited Cutaneous
SSc. The typical patient with limited cutaneous SSc is a female (or
a male who has worked with vibrating machines, plastics, or in mining),
aged 30 to 50, who presents with a 10- to 15-year history of numbness
and a "dead" or "wooden" sensation associated with color changes (often
pallor only) of, at first, the second and third fingers of the dominant
hand. Full-fledged Raynaud's phenomenon usually develops symmetrically
in both hands, fingers 2 through 5, with increasing frequency, especially
in winter; there may be a history of hard crusting lesions on the fingertips,
initially healing in warm weather. General stamina may be decreased,
and there may be breathlessness with minimal exertion (see Table 437-2).
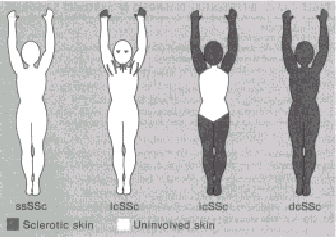
FIGURE 437-1. A pictorial representation of skin involvement
in systemic sclerosis. Note that the limited cutaneous SSc patient (Table
437~. 2) may have subtle thickening of eyelid, neckfold, and armpit
s in. Abbreviations: ssSSc = systemic sclerosis sine scleroderma; IcSSc
= limited cutaneous systemic sclerosis; icSSc = intermediate cutaneous
systemic sclerosis; dcSSc = diffuse cutaneous systemic sclerosis. (Reprinted
with permission from Giordano M, et al.: J Rheumatol 13:911, 1986.)
DIAGNOSIS. The annual
incidence of Raynaud's phenomenon is substantially greater than the
incidence of all diffuse connective tissue syndromes combined (Table
437-4). To select those Raynaud's patients destined to develop scleroderma
and related disorders when a careful history and physical examination
reveal no features of connective tissue disease, including no signs
of peripheral ischernia, the single best test is widefield nailfold
capillaroscopy-a noninvasive, reproducible, cost-effective, permanent
identification of the connective tissue disease-prone patient. Coupled
with autoimmune serology and possibly a test of vascular injury or platelet
activation/release (such as plasma beta- thromboglobulin levels), capillary
examination is an important procedure in the early detection of connective
tissue disease.
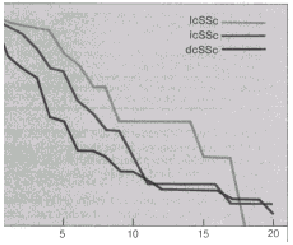
FIGURE 437-2. The cumulative survival rate (CSR, age-adjusted
survival) in per cent plotted against time in years for diffuse cutaneous
SSc and limited cutaneous SSc (crest syndrome) patients, showing the
substantially reduced survival of diffuse cutaneous SSc patients (Table
437-2). An intermediate group with both intermediate survival and extent
of skin involvement has been identified by Giordano et al. For abbreviations,
see Figure 437-1. (Reprinted with permission from Giordano M, et al.:
J Rheumatol 13:911, 1986.)
The diagnosis of diffuse
SSc is straightforward. A previously well person is now sick with the
triad of Raynaud's phenomenon, nonpitting edema, and hidebound skin
that may eventually cover virtually the entire body, sparing only the
back and buttocks. There are very few alternative diagnoses that must
be seriously entertained. Other causes of Raynaud's phenomenon are not
usually accompanied by edema, and other causes of edema are not usually
associated with Raynaud's phenomenon. The key questions in such patients
are (1) Are features of other connective tissue diseases present? and
(2) What internal organs are affected?
If symmetric, erosive polyarthritis
is present, an overlap between SSc and rheumatoid arthritis should be
considered; if fever and a characteristic malar rash are present, overlap
with systemic lupus erythernatosus is likely. Most often these features
are not present, and the second question represents the primary focus.
Each visceral target organ (esophagus, lungs, kidneys, heart) of SSc
deserves screening.
Abnormal skin texture provides
the definitive diagnostic criterion of SSc in over 90 per cent of patients.
When distal to the metacarpophalangeal (MCP) joints only, it is called
sclerodactyly and is not diagnostic of SSc. Firm, taut, hidebound skin
proximal to the MCP joints represents the major diagnostic criterion.
Skin biopsy is usually not more sensitive diagnostically than the experienced
touch. Skin changes also distinguish the three prognostically different
subsets. If truncal skin changes are present, the patient has diffuse
cutaneous SSc, and close surveillance of visceral function is indicated.
If skin changes are limited to the hands, fingers, and face, limited
cutaneous SSc is present, yearly evaluation is adequate, and management
should focus on the Raynaud's phenomenon. If the skin is of normal texture,
two possibilities are suggested: the patient formerly had abnormal skin
changes-either diffuse or limited cutaneous-which have subsided (regressive
systemic sclerosis); or the patient has visceral disease in the absence
of skin changes which occurs in at least 5 per cent of SSc patients
(Table 437-2, SSc sine scleroderma).
DIFFERENTIAL DIAGNOSIS.
Connective tissue disorders are constellations of organ system involvements
(skin, lungs, intestinal tract, serosal surfaces, joints, heart, central
nervous system); each system involved can present immune-inflammatory,
proliferative-erosive, or fibrotic-atrophic-insufficiency changes at
different stages of the syndrome or disorder. Virtually none of the
systems involved or the stages of involvement of those systems are entirely
specific for the particular syndrome or disorder. It is not surprising,
therefore, that the nomenclature is confusing. Terms such as mixed connective
tissue disease (MCTD), undifferentiated connective tissue syndromes
(UCTS), and overlap syndromes have emerged to describe the same patients.
MCTD was introduced to describe
anew the already wellknown overlap patients with features of myositis,
lupus, and scleroderma. The early features of such patients were largely
inflammatory and therefore briefly responsive to glucocorticoid therapy;
proliferative and fibrotic features were not responsive to treatment.
Neither the clinical syndromes, the laboratory tests proposed (extractable
nuclear antigen [ENAJ antibodies to ribonucleoprotein [RNPI), nor the
response to therapy was specific. Therefore, the introduction of MCTD
provided no new understanding beyond the time-honored overlap syndrome,
which remains the preferred term for established, stable connective
tissue disorders with features of more than one traditional disorder
(such as rheumatoid arthritis-lupus overlap). In the early patient with
inflammatory or edematous features that are insufficient for an established
diagnosis, the term undifferentiated connective tissue syndrome (UCTS)
is preferred. Some use UCTD here as a hybrid acronym.
Eosinophilic fasciitis
is a syndrome that, when acute, is distinct from scleroderma and that
blends into the scleroderma spectrum of disorders in its chronic form.
Young, vigorous persons note, often after strenuous exertion, the onset
of swelling and tautness of the skin of the trunk and proximal extremities
with a brawny texture that may be tender. Raynaud's phenomenon is usually
absent, and the hands and feet are usually spared. Initially, visceral
disease is absent. Deep skin and subcutaneous biopsies show inflammatory
changes including the deep fascia, the subcutis, and the dermis. Eosinophilia.
is present and eosinophils may or may not be present in the skin lesions.
Symptoms subside with glucocorticoid therapy and also with no therapy
over time , Eosinophilic fasciitis has been associated with aplastic
anemia. In a substantial proportion of chronic patients, the visceral
involvement of systemic sclerosis has been documented.
CLINICAL
MANIFESTATIONS.
Peripheral Vascular System.
Pallor is the most definitive of the triphasic responses of Raynaud's
phenomenon. In the presence of constant or episodic cyanosis alone,
the diagnosis should be suspect. The circumstances that provoke pallor
and the "dead" sensation of the fingers are usually reproducible in
the individual patient (handling cold or frozen items, emotional disturbances).
Persistent Raynaud's attacks may lead to a webbing phenomenon (like
the frenulum of the tongue) binding the finger nail to the finger tip
skin of involved fingers. This is evidence of structural vascular and
persistent ischernic disease, as are the more obvious finger tip calluses,
digital ulcerations (of finger tips or over dorsal proximal interphalangeal
joints), overt ischernic tissue, or calcification; the toes, the nose,
and the ears may be affected in Raynaud's attacks as well. The more
widespread the areas involved, the more likely is systemic disease.
The Skin. The skin
is the most distinctive diagnostic feature of SSc; the diagnosis can
be made unequivocally by the texture and location of hidebound skin.
In patients with diffuse SSC, skin tautness can limit movement at the
wrists, elbows, shoulders, mouth, and thorax (less frequently the hips,
knees, and ankles). When fully hidebound, the skin appears to become
paper-thin over points of bony protrusion, such as the proximal interphalangeal
joints, the u1nar styloid process, the olecranon process, the bridge
of the nose, and the cheekbones. Gentle pressure over these areas removes
all blood from the capillaries; the refilling time can be used as a
rough approximation of the degree of ischernia and the propensity to
ulcerate.
Gastrointestinal System.
If sensitive diagnostic techniques are used, esophageal hypornotility,
by far the most common manifestation of gastrointestinal SSc, can be
documented in over 90 per cent of patients with both diffuse and limited
cutaneous SSc. Many patients do not notice the subtle symptoms of esophageal
SSc, which include a vertical substernal burning pain particularly at
night, the occasional sense that a pill or large bit of meat "has not
gone all the way down," or "heartburn" on lying down soon after a full
meal. The single best screening test for esophageal hypornotility is
the radionuclide esophageal transit time; it is noninvasive, safe, and
can be relied upon when negative. Because severe esophageal complications,
including stricture, can be prevented by early management and because
intestinal involvement with SSc does not occur without esophageal involvement,
the esophagus of all patients suspected of SSc should be examined for
hypornotility. Patients with slow transit times should be further studied
with both barium swallow (using light barium and the recumbent position)
to detect structural abnormalities (hiatus hernia), and with esophageal
motility studies, the definitive procedure for esophageal SSc. The earliest
detectable abnormality is a reduction in resting lower esophageal sphincter
(LES) pressure, which may be an isolated early finding or may be associated
with reduced smooth muscle contraction (secondary and tertiary waves)
of the distal two thirds of the esophagus. Upper third, striated muscle
dysfunction suggests an overlap syndrome with dermatomyositis. If peptic
esophagitis with mucosal ulceration is well-established, LES pressure
may be increased and the diagnosis of achalasia could be incorrectly
entertained. The presence of other features of SSc and reduced LES pressure
after treatment are helpful.
Gastric hypornotility may
be present but is not often of clinical significance. Small intestinal
hypornotility, determined by upper GI series with small bowel follow-through,
occurs in 10 to 20 per cent of patients, all of whom have esophageal
hypomotility; these may occur in the absence of cutaneous scleroderma.
It need not be searched for in the asymptomatic patient because it consistently
declares its presence by postprandial bloating, abdominal distention
with diffuse pain, intermittent diarrhea with or without steatorrhea,
and weight loss from malabsorption. Abdominal attacks with adynamic
ileus may mimic mechanical obstruction and lead to surgical intervention,
from which some patients recover poorly and slowly, if at all.
Pulmonary System.
Although renal failure was formerly the major threat to life in SSc,
the combined impact of several pulmonary abnormalities now seems to
be the number one cause of fatal involvement in this disease. Pleurisy
and pleural effusions, pulmonary hypertension, interstitial lung disease
with fibrosis, and ultimately obstructive pulmonary disease all may
be a part of pulmonary SSc. Because the patient is often sedentary from
skin or joint restrictions, shortness of breath or dyspnea on exertion
are surprisingly late complaints; also, standard chest roentgenography
is not a sensitive screening procedure. More than half of SSc patients
selected for the absence of pulmonary symptoms and for a normal chest
radiograph show reproducible abnormalities on pulmonary function tests.
Patients who smoke show a much higher positive proportion. The single
breath diffusion capacity, which measures the balance between ventilation
and perfusion, is a sensitive pulmonary screening tool. Mild reductions
in vital capacity are common as well. In smokers, there may be evidence
of small airway obstruction. Pleural effusions are usually silent and
bland. They take on clinical significance primarily in the patient with
established restrictive lung disease (decreased vital capacity) in whom
the aspiration of an effusion may improve ventilation. They are present
in two thirds of patients at postmortem examination. In the immunosuppressed
patient, infection may present with 11silent- empyerna. Pulmonary hypertension
may be sudden in onset and constitutes a medical emergency. All patients
with SSc should be followed closely for changes in the second heart
sound over the pulmonic area and for the pulmonic valve closure component
of that second sound, detected by the splitting of S2 on deep inspiration.
The appearance of tricuspid regurgitation or right ventricular enlargement
is evidence of established pulmonary hypertension. The chest x-ray may
provide evidence of enlarged pulmonary arteries but often does not;
the echocardiogram, while key in detecting cardiac SSc, has been disappointing
in detecting pulmonary hypertension. Doppler techniques measuring tricuspid
insufficiency (present in most patients with increased right ventricular
pressures) are promising in the early detection of pulmonary hypertension.
Aggressive attempts to lower pulmonary artery pressure should be instituted
Renal System. At one
time, the abrupt onset of accelerated hypertension and oliguria ("scleroderma
renal crisis") accounted for the majority of deaths in SSc. Fortunately,
with early identification and treatment with inhibitors of angiotensin-converting
enzymes (captopril, enalapril), the incidence of renal involvement and
its consequences have been greatly improved. All patients fulfilling
the criteria for diffuse SSc should be suspect for renal involvement
and should be followed with 24-hour urine collections for protein excretion
and creatinine clearance three to four times a year. Excretion of greater
than 750 ing of protein per 24 hours or clearances of less than 60 ml
per minute, or distinct changes in either proteinuria or glomerular
filtration rate (GFR), should initiate measurements of resting renin
and, if elevated, treatment. Increases in blood pressure, pulse rate,
accelerated increases in edematous skin tightening (rapidly increasing
skin score), or the appearance of microangiopathic hemolytic anemia
or disseminated intravascular coagulation (see Ch. 167) may also herald
the onset of renal involvement. The sudden appearance of renal SSc when
other features of the disease appear more indolent is a characteristic
of the kidney's unique ability to autoregulate its own blood flow. The
typical small artery lesion of intimal proliferation develops slowly
in the kidney of SSc patients with gradual reduction in renal blood
flow, until both renal and glomerular flow drop abruptly and renal failure
ensues. A rapid acceleration of these changes is associated with the
onset of hyper-reninemia. It is during this accelerated phase that hypertension,
funduscopic vascular changes (hemorrhages and exudates), and microangiopathic
hemolytic anemia appear. The key to successful management of renal SSc
is to identify the population at risk (those with diffuse SSc), to detect
declining GFR early, and to treat expectantly. One remarkable feature
of renal SSc is the ability of some patients to regain renal function
after months to years (up to four years) of end-stage renal disease
and hemodialysis. Very little is known of the mechanisms of this slow
reparative process. Whatever the reason, it stays the-hand that would
undertake nephrectomy.
Cardiac System. More
than 90 per cent of patients with diffuse SSc (truncal skin involvement)
have some form of cardiac involvement. Rarely, acute pericarditis with
a friction rub is present; more frequently, a silent pericardial effusion
appears slowly with ankle edema and shortness of breath as the presenting
features. Echocardiography is the diagnostic procedure of choice. Pericardial
effusions may predispose to renal failure by unknown mechanisms. By
electrocardiographic monitoring and electrophysiologic studies, 80 per
cent of diffuse SSc patients without cardiovascular symptoms have evidence
of cardiac involvement; in more recent studies, 95 per cent show abnormalities
of thallium reperfusion. Intermittent myocardial ischernia with the
acute pathologic concomitant of contraction band necrosis seems to precede
fibrosis, suggesting that spasm of the intramyocardial vessels plays
a role in cardiac SSc. Most, but not all, of these patients have normal
coronary arteries by coronary angiography.
Articular and Musculoskeletal.
Approximately 10 per cent of SSc patients present with a symmetric small
joint polyarticular synovitis indistinguishable from rheumatoid arthritis.
Within a year, the pattern changes abruptly with subsidence of joint
complaints and the appearance of Raynaud's phenomenon, edema, and diffuse
cutaneous SSc. The presence of scleroderma-pattern nailfold capillary
changes and a positive antinuclear antibody pattern can identify these
patients during their polyarticular phase, prior to the development
of cutaneous changes, as destined to develop SSc. About one half of
SSc patients develop stiffness and swelling of the fingers, wrists,
knees, and ankles, concomitant with cutaneous changes. Morning stiffness
may be present. Signs of inflammation are usually mild. Polymorphonuclear
leukocytes are usually present in synovial fluid. On biopsy, the synovium
is mildly inflamed, with a distinctive deposition of fibrin throughout
the synovium. Obliterative microvascular disease and diffuse fibrotic
changes occur at a later stage.
Indolent myopathy
is common in SSc. It is difficult to distinguish from atrophy caused
by taut skin. Most patients show diffuse atrophy of the extremities
with slight elevations of muscle enzyme levels (CPK and aldolase); these
features are refractory to glucocorticoids or to immunosuppressive therapy.
Mild myositis of SSc is best left untreated. Less frequently, abrupt
proximal muscle weakness develops and is associated with 10- to 50-fold
increases in muscle enzymes, electromyographic features of acute myositis,
and lymphoid cell infiltration with muscle fiber necrosis on biopsy.
These patients generated the initial confusion regarding mixed, overlap,
or undifferentiated connective tissue syndromes; they usually respond
to glucocorticoid therapy. Other. In the second and third decades following
the onset of Raynaud's phenomenon, a small but significant proportion
of patients with limited cutaneous SSc develop unilateral or bilateral
trigerninal neuralgia, which can be disabling. An increasing number
of male SSc patients, especially those with diffuse disease, experience
impotence after one or two years of the disease. Impotence is thought
to have an organic neurovascular cause, because of diminished or absent
nocturnal tumescence. It is refractory to treatment.
Dry eyes (keratoconjunctivitis
sicca), dry mouth (xerostomia), or both occur in approximately one
fourth of SSc patients. Salivary gland biopsies may show mononuclear
cell infiltrates or replacement fibrosis. Supportive care with secretion
substitution (artificial tears) and stimulation (lemon candy) provides
some relief.
TREATMENT.
No therapy has been shown to halt the progression of cutaneous or visceral
SSc in a controlled, prospective study.(note:Since
this was written, antibiotics have been shown to put the disease in
remission. Members of this support group also can testify to the reversal
and remission of the disease with the use of the Antibiotic Protocal)
A major source of confusion in assessing therapy is the
dependence on softening skin as a key outcome measurement and the natural
tendency for hidebound skin to soften after several years (dubbed regressive
systemic sclerosis). Skin changes cannot be used as indications of the
lessening of the vascular and microvascular disease. The most distinctive
change in the natural history of diffuse cutaneous SSc in the last decade
has been the reduction in the proportion of patients who develop renal
failure. This change has occurred with the advent of more powerful agents
to control the accelerated hypertensive phase of renal failure, especially
inhibitors of the angiotensin-converting enzyme, captopril and enalapril.
Indications for immediate treatment are hypertension (an increase of
30 mm Hg systolic or 15 mm Hg diastolic blood pressure, no matter what
the absolute level); a reduction in creatinine clearance of 30 ml per
minute or to a clearance below 60 ml per minute; and microangiopathic
anemia. If the serum creatinine value is less than 4.0 mg per deciliter,
the crisis of renal scleroderma can often, but not always, be averted.
Continued intensive treatment is indicated even if hemodialysis is instituted,
since some patients can regain function sufficient to obviate dialysis
after as long as four to five years. D-Penicillamine* has been strongly
advocated on the basis of retrospective studies that showed skin softening
after two years. The proportion of patients who develop significant
side effects is 30 to 40 per cent. It is a difficult drug to tolerate.
Colchicine* has also been proposed as capable of influencing cutaneous
changes in SSc. Brief cross-over studies were inconclusive, and longer
open studies were promising but uncontrolled. Colchicine is better tolerated
than D-penicillamine. Glucocorticoids in moderate doses (30 to 40 mg
per day in divided daily doses) effectively reduce the inflammatory
and edematous changes in SSc but have no effect on the fibrotic features.
When pulmonary SSc can be shown to have an active inflammatory component
by bronchoalveolar lavage or gallium/indium scans, a brief trial of
high-dose (60 to 80 mg in divided daily doses) glucocorticoids is indicated.
Also it may help to reduce pulmonary hypertension if used early in the
course of its development, usually in conjunction with calcium channel
blockers.
The management of Raynaud's
phenomenon has improved in recent years (see Ch. 57); nonetheless, even
the most successful management of the vasoactive features of SSc does
not appear to slow or stop the continuing appearance of new fibrotic
or visceral manifestations. Sometimes a change in life style is sufficient.
Clothing should protect the trunk to encourage heat dissipation via
peripheral vasodilatation. Extremes of cold, exhaustion, or stress should
be avoided. Nitroglycerin ointment applied locally along the course
of the digital arteries to those fingers showing severe ischemia is
helpful. Selective sympathetic blockade, especially postganglionic alpha
blockade with prazosin, usually reduces symptoms but may be difficult
to tolerate due to palpitation and orthostatic hypotension. Inhibitors
of the slow calcium channels of cell membranes have been a significant
advance in the management of Raynaud's phenomenon. At present, nifedipine
in gradually increasing doses is popular, but verapamil and diltiazem
have their proponents as well. When tissue necrosis is present (gangrene),
prompt hospital admission for stellate ganglion blockade or epidural
blocks is indicated. As an example of how therapeutic trials in SSc
must be conducted to provide meaningful results in this indolent, variable
disorder, the negative trial of chlorambucil by Furst and colleagues
is cited. The agent was not better than placebo. Fifty-two patients,
with a mean of 7.2 years of symptoms, were treated for three years each.
Extensive inpatient evaluation of involvement was carried out at 6,
12, 24, and 36 months, evaluating skin, skeletal, pulmonary, cardiac
(left and right heart), renal, upper and lower gastrointestinal, muscular,
and global involvement. "Slope estimates" for each patient and each
organ system were constructed and compared. It is a testimony to how
much more we need to understand about SSc that a drug which has been
as carefully evaluated as was chlorambucil by Furst et al., has been
shown not to change the course of the disease.
|



